Article Text

Abstract
Background Relatives of patients with familial interstitial pneumonia (FIP) are at increased risk for pulmonary fibrosis. We assessed the prevalence and risk factors for preclinical pulmonary fibrosis (PrePF) in first-degree relatives of patients with FIP and determined the utility of deep learning in detecting PrePF on CT.
Methods First-degree relatives of patients with FIP over 40 years of age who believed themselves to be unaffected by pulmonary fibrosis underwent CT scans of the chest. Images were visually reviewed, and a deep learning algorithm was used to quantify lung fibrosis. Genotyping for common idiopathic pulmonary fibrosis risk variants in MUC5B and TERT was performed.
Findings In 494 relatives of patients with FIP from 263 families of patients with FIP, the prevalence of PrePF on visual CT evaluation was 15.6% (95% CI 12.6 to 19.0). Compared with visual CT evaluation, deep learning quantitative CT analysis had 84% sensitivity (95% CI 0.72 to 0.89) and 86% sensitivity (95% CI 0.83 to 0.89) for discriminating subjects with visual PrePF diagnosis. Subjects with PrePF were older (65.9, SD 10.1 years) than subjects without fibrosis (55.8 SD 8.7 years), more likely to be male (49% vs 37%), more likely to have smoked (44% vs 27%) and more likely to have the MUC5B promoter variant rs35705950 (minor allele frequency 0.29 vs 0.21). MUC5B variant carriers had higher quantitative CT fibrosis scores (mean difference of 0.36%), a difference that remains significant when controlling for age and sex.
Interpretation PrePF is common in relatives of patients with FIP. Its prevalence increases with age and the presence of a common MUC5B promoter variant. Quantitative CT analysis can detect these imaging abnormalities.
- Idiopathic pulmonary fibrosis
- Interstitial Fibrosis
- Imaging/CT MRI etc
Statistics from Altmetric.com
Key messages
What is the key question?
What are the risk factors for undiagnosed pulmonary fibrosis in first-degree relatives of patients with familial interstitial pneumonia (FIP) and can deep learning methods be used to detect it?
What is the bottom line?
Undiagnosed pulmonary fibrosis in first-degree relatives of patients with FIP is common, associated with the MUC5B promoter variant, and deep learning methods can be used to detect it.
Why read on?
Our article describes in detail the radiological findings of early pulmonary fibrosis in at-risk subjects, genetic and clinical risk factors for it, and the application of a deep learning algorithm on CT imaging of these subjects.
Introduction
Idiopathic pulmonary fibrosis (IPF), the most common idiopathic interstitial pneumonia (IIP), is a poorly understood disease characterised by progressive lung parenchymal scarring, impaired gas exchange, loss of lung function, physical debilitation and shortened life span. Median survival is approximately 3 years from the time of diagnosis,1 and the clinical course is unpredictable.1 There are no curative therapies2 3 other than lung transplantation.
Recent studies have identified genetic variants, both common and rare, associated with both familial and sporadic forms of pulmonary fibrosis.4–7 A MUC5B promoter variant has been shown to be the most important variant associated with familial and sporadic disease and with interstitial lung abnormalities (ILAs).4 5 8 Numerous rare variants in TERT (and other telomerase-pathway genes) have been thought to be critical in familial diseases6 9–11 ; more recently, a common variant in TERT 5 has been associated with sporadic and familial disease.
Better understanding and recognition of early pulmonary fibrosis is critical because medical therapies have been shown to slow progression, not to reverse existing fibrosis; intervention before irreversible fibrosis has become extensive and has the potential to improve quality of life and to decrease morbidity. While IPF affects approximately five million people worldwide,1 1.8% of the general population and 14% of the familial at-risk population aged ≥50 years have radiological findings of undiagnosed pulmonary fibrosis.8 12 13 Large cohort studies indicate that ILAs, postulated to represent early pulmonary fibrosis, are associated with increased mortality and generally progress over time.12 13 Members of families with two or more cases of pulmonary fibrosis (familial interstitial pneumonia (FIP)) have been identified as an ‘at-risk’ population. In a previous study of relatives of patients with FIP, 14% had ILAs on high-resolution CT (HRCT), and 35% had an abnormal transbronchial biopsy indicating interstitial lung disease (ILD).14
HRCT plays a key role in the diagnosis of the IIPs, including IPF. Currently, visual pattern diagnosis by thoracic radiologists, in conjunction with multidisciplinary clinical conference, is the gold standard for diagnosing IIPs.15 However, visual assessment is imprecise and hampered by interobserver variation.16 Quantitative high-resolution CT (qHRCT) evaluation provides measures of fibrosis extent that, in subjects diagnosed with IPF, correlate with degree of physiological impairment at baseline and may be more sensitive to subtle changes in disease status than routinely used physiological metrics.17 18 The design and utility of qHRCT methods in the context of early forms of fibrotic ILD requires further study.19 Deep learning methods have been increasingly used in imaging to identify and classify CT patterns20 and may be valuable in detection of early lung fibrosis.
A key strength of deep learning algorithms, such as convolutional neural networks (CNNs), is that they simultaneously optimise feature extraction and calculation of classification rules. During training, CNNs ‘learn’ to extract the most effective image features, including textural features at multiple scales, for the given classification task. This is in contrast to more traditional methods that rely on separate processes to engineer and select features, then develop classification rules. Engineered features are designed manually, often by using combinations of standard statistical or image processing calculations, and may not be the most effective features for a given classification task like the discrimination of pulmonary fibrosis.
This study aimed (1) to examine risk factors, including two common fibrosis-associated genetic variants in MUC5B and TERT, for undiagnosed preclinical pulmonary fibrosis (PrePF) in first-degree relatives of patients with FIP; and (2) to determine the utility of a deep learning, texture-based qHRCT method in the detection of PrePF in this at-risk cohort.
Materials and methods
Screening of relatives of patients with FIP
At the University of Colorado, National Jewish Health and Vanderbilt University (COMIRB #15–1147, NJH IRB 1441a and Vanderbilt IRB #020343), non-Hispanic white (NHW), first-degree relatives of patients with FIP, defined as those in families with two or more cases of pulmonary fibrosis (online supplementary figure S1), were contacted. After informed consent, first-degree relatives greater than 40 years of age without a known diagnosis of pulmonary fibrosis were offered HRCT scans of the chest and peripheral blood draw. Those younger than 40 years of age or who reported on prescan questionnaires to be personally affected by pulmonary fibrosis were excluded (figure 1).
Supplemental material
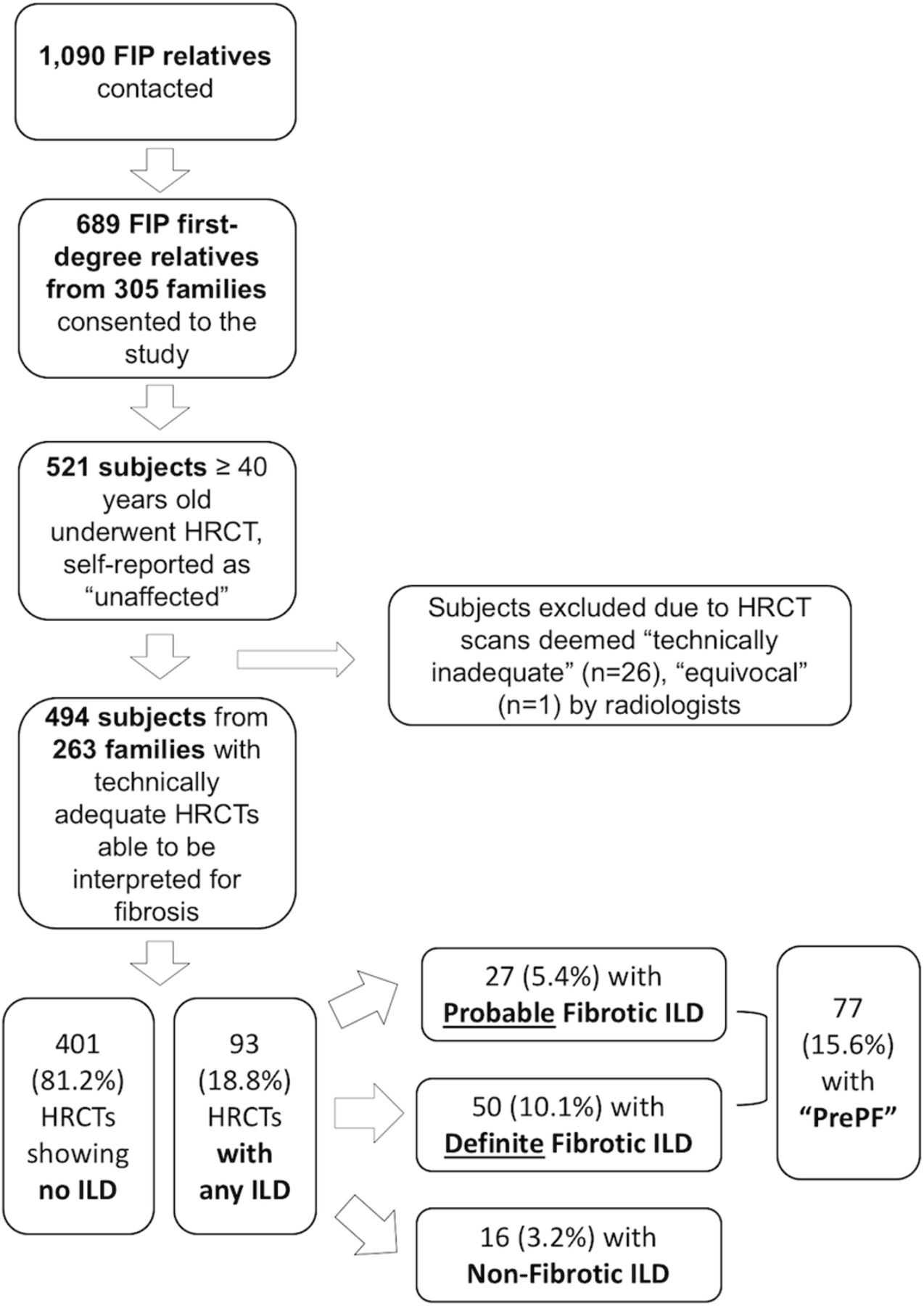
Enrolment and screening flowchart description of enrolment process and results for study subjects. FIP, familial interstitial pneumonia; HRCT, high-resolution CT; ILD, interstitial lung disease; PF, pulmonary fibrosis.
Visual CT review
See online supplement for details (online supplementary file 1). HRCT scans were interpreted by study radiologists using a standardised method.21 ‘PrePF’ was defined as the presence of ‘probable’ or ‘definite’ fibrotic ILD on HRCT in relatives of patients with FIP who had no known diagnosis of pulmonary fibrosis at the time of study enrolment (figures 1 and 2).
Supplemental material

Representative images from cohort subjects. (A) HRCT image of the chest from a study subject whose scan was read as normal, without signs of ILD or fibrosis. (B) HRCT image from a subject who was categorised as having ‘probable fibrotic ILD’. (C) Rrepresentative HRCT image from a subject who was characterised as having ‘definite fibrotic ILD’. (D) HRCT image from a case of previously diagnosed, established idiopathic pulmonary fibrosis in one of the study families. HRCT, high-resolution CT; ILD, interstitial lung disease.
Quantitative CT
Inspiratory HRCT series with a slice thickness of ≤1.25 mm and spacing of ≤20.0 mm were selected for quantitative analysis. This included 212 volumetric series with thin, contiguous sections (slice thickness and spacing both ≤1.25 mm) and 191 non-volumetric scans (56 with slice spacing of >1.25 and <10 mm, 65 with slice spacing of 10 mm and 70 with slice spacing of 20 mm). Technically inadequate scans were omitted (online supplementary file S2). In addition, 100 inspiratory volumetric HRCTs of normal, never-smoker control subjects from the COPDGene cohort were analysed (online supplementary table S1).22 23 In an initial process, the lungs were segmented using a deep learning model that had been trained using CTs of subjects with and without fibrosis. Details are available in the online supplement (online supplementary file 1). Trained analysts verified lung segmentation visually and made edits, if necessary. Examples of the categorisation of different parts of CT scans are shown in figure 3. Some studies were acquired with contiguous thin axial sections, while others used 1 or 2 cm intervals. Reconstruction kernel, a parameter that affects image sharpness and noise, was not standardised.
Supplemental material

Categorisation of regions of HRCT images using quantitative methodology representative axial HRCT images visually assessed as ‘no fibrosis’ (A), ‘probable fibrotic ILD’ (B) and ‘definite fibrotic ILD’ (C). Below each is the corresponding quantitative HRCT results for the above scan; regions classified as fibrotic are shown in red. (A) ‘No fibrosis’ fibrosis extent 0.10% (log(fibrosis score)=−2.30); (B) ‘probable fibrotic ILD’ fibrosis extent 12.46% (log(fibrosis score)=2.52); (C) ‘definite fibrotic ILD’ fibrosis extent 24.05% (log(fibrosis score) 3.18). HRCT, high-resolution CT; ILD, interstitial lung disease.
Fibrosis quantification on CT scans was performed using a second deep learning technique, called deepDTA, consisting of a CNN algorithm trained with image regions of normal and abnormal lung identified by expert radiologists. Training data and an earlier algorithm version, called data-driven textural analysis, were described previously.17 Here, a more complex CNN architecture was employed that classifies image regions using pixel and texture features extracted by multiple convolutional layers at different scales. The CNN classifies image regions as either normal or fibrotic, with the fibrotic category trained using image regions labelled by a radiologist as reticular abnormality, honeycombing or traction bronchiectasis. Subject-level HRCT fibrosis scores were computed as the percentage of total lung volume classified as fibrotic (figure 3 and online supplementary figure S3). A simpler previously described densitometric analysis of HRCTs, per cent high-attenuation area (%HAA), was also performed for comparison24 (see online supplement).
Supplemental material
Blood Processing, Genotyping, and Autoantibody Testing: See online supplement (online supplementary file 1).
Statistical analysis
Analysis of the effect of specific alleles on PrePF risk was performed using minor allele frequency (MAF) for comparison of variant prevalence in the study groups; statistical significance was determined with a z-score test for proportions or a mixed effects logistic regression model when controlling for other variables (age, sex, smoking history and family (random effect)) in dominant and log-additive models.
Distribution of qHRCT fibrosis scores was left skewed, as was %HAA, so these values were log transformed prior to analyses (online supplementary figures 5–8). Log of qHRCT fibrosis score (hereafter, ‘fibrosis score’) and log (%HAA) were compared with visual scores using analysis of variance and Tukey’s honestly significant difference test. To determine the ability of qHRCT scores to predict visual diagnosis of PrePF, receiver operating characteristic (ROC) analysis was performed. Optimal threshold for discriminating visual diagnosis of fibrotic ILD was determined with Youden’s method. Fivefold cross-validation was performed to test detection accuracy, sensitivity and specificity, and consistency of optimal threshold. Linear regression was performed to test the association between the MUC5B genotype and qHRCT fibrosis score and log (%HAA).
A p value of <0.05 was considered statistically significant for differences between groups, as well as for associations between individual variables and outcomes in linear and logistic regression modelling. Statistical analyses were performed using RStudio (V.0.99.473).
Results
Study cohort characteristics
A total of 1090 first-degree relatives of patients with FIP were contacted; 521 eligible subjects underwent HRCT screening (figure 1). Of the 521 subjects, 26 were excluded due to technical inadequacy of images and 1 for an equivocal consensus read by study radiologists. The remaining 494 subjects from 263 families were included in the analyses. Subjects’ mean age was 57 years (SD 9.6), 189 (38%) were male and 148 (30%) were either current or former smokers. The minor allele (T) frequency of the MUC5B promoter variant rs35705950 was 0.22 in this cohort; 42% of the subjects in this cohort had one or two copies of the minor allele (table 1). The minor allele (C) frequency of the TERT variant rs2736100 was 0.47 in the entire cohort; 69% of the subjects in the cohort had one or two copies of the minor allele (table 1).
Screening cohort subject characteristics
Prevalence of PrePF in relatives of patients wih FIP
Of the 494 HRCT scans, 399 showed no CT evidence of ILD, and 93 showed evidence of ILD, either fibrotic (27 probable and 50 definite) or non-fibrotic (n=16). Therefore, among these 494 subjects who reported being personally unaffected by pulmonary fibrosis, the PrePF prevalence was 15.6% (n=77) (figure 1).
The CT patterns noted in visually identified PrePF subjects (table 2) show that a possible, probable or definite usual interstitial pneumonia (UIP) pattern was the most commonly considered (n=59, 77% of all PrePF cases). Nonspecific interstitial pneumonia (NSIP) was considered in 45 subjects (58% of all PrePF cases). The fibrotic changes were most commonly lower-lobe predominant and subpleural in nature, consistent with a UIP pattern (table 2). Non-fibrotic ILD scans, on the other hand, generally had more diffuse, upper-lobe predominant abnormalities (online supplementary table S2 and S3).
Visually identified patterns of CT abnormalities in scans with probable or definite fibrotic ILD
There were 402 study subjects with HRCT scans that were technically adequate for quantitative assessment (online supplementary figure S2). Two hundred twelve of the scans had both slice thickness and spacing of ≤1.25 mm (thin, contiguous); of the remaining 191 scans, 56 had slice spacing >1.25 and <10 mm, 65 had a slice spacing of 10 mm, and 70 had a slice spacing of 20 mm. Volumetric HRCT scans on an additional 100 COPDGene subjects were included as normal controls (online supplementary table S1 and figure S3). HRCT CNN fibrosis score means were significantly different (p<0.0001) across groups defined by visual diagnosis (figure 4). Comparison of means showed fibrosis scores were significantly different comparing each group (all between-group comparisons p<0.01). The means of log (%HAA) scores were also significantly different across visual scoring groups (p<0.0001), and individual between-group comparisons showed log (%HAA) was significantly different in most comparisons (p<0.0001), except between the probable and definite visual scores (p=0.35, online supplementary figure S7).
Supplemental material

Fibrosis score by visual diagnosis. Boxplots of fibrosis scores based on quantitative HRCT assessment for each visual diagnosis category. Fibrosis score means were significantly different (analysis of variance, p<0.0001) across groups defined by visual diagnosis. Comparison of fibrosis score between groups showed significant differences for all individual comparisons (p<0.01 for all). ILD, interstitial lung disease.
ROC analyses showed that the fibrosis score discriminates subjects with visual diagnosis of PrePF (figure 5B). The average area under the curve (AUC) in the fivefold cross validation was 0.92 (range 0.91–0.93) and average accuracy, sensitivity and specificity in the test partitions were 0.85 (range 0.81–0.88), 0.81 (range 0.71–0.92) and 0.86 (range 0.79–0.90), respectively. Optimal threshold for log fibrosis score was 0.60 (range 0.53–0.71), corresponding to 1.8% fibrotic area in the examined lung. Using a cut-off of 0.60 for log fibrosis score on the entire dataset, the sensitivity was 84% (95% CI 72% to 92%), the specificity was 86% (95% CI 83% to 89%) and the accuracy was 86%, while the positive predictive value of this test was only 46% (95% CI 36% to 55%), and the negative predictive value was 97% (95% CI 95% to 99%) (figure 5B,C).

ROC curves for quantitative imaging measures of fibrosis and PrePF. (A) ROC curves for visual diagnosis compared with log per cent high-attenuation area. For this quantitative method, the mean AUC was 0.80 (range 0.79–0.81). (B) ROC curves for visual diagnosis compared with fibrosis scores. ROC analysis showed that the fibrosis score discriminates subjects with visual diagnosis of PrePF. Average AUC in fivefold cross validation was 0.92 (range 0.91–0.93), and average accuracy, sensitivity and specificity in the test partitions were 0.85 (range 0.81–0.88), 0.81 (range 0.71–0.92) and 0.86 (range 0.79–0.90), respectively. The optimal threshold for log fibrosis score was 0.60 (range 0.53–0.71), corresponding to 1.8% fibrotic area in the examined lung. (C) Density plots of fibrosis scores for visually diagnosed PrePF (pink) and no fibrosis (blue) scans—the fibrosis score optimal threshold is indicated with the red line (0.60). AUC, area under the curve; HRCT, high-resolution CT; PF, PrePF, preclinical pulmonary fibrosis; ROC, receiver operating characteristic.
Compared with the classification achieved with the CNN as described earlier, ROC analysis of log %HAA had a lower mean AUC of 0.80 (range 0.79–0.81) and average accuracy, sensitivity and specificity of 0.67 (range 0.63–0.70), 0.82 (range 0.75–0.91) and 0.64 (range 0.62–0.70), respectively (figure 5A). The mean optimal threshold for log %HAA ranged from 1.49 to 1.57. Using a cut-off of 1.49 for log %HAA, the sensitivity was 89% (95% CI 78% to 95%), the specificity was 62% (95% CI 57% to 66%) and accuracy was 60%, while the positive predictive value of this test was only 24% (95% CI 19% to 30%), and the negative predictive value of this test was 96% (95% CI 95% to 99%).
Risk factors for PrePF
Subjects with PrePF were older (mean age 65.9 years, SD 10.1) than those without fibrosis (mean age 55.8, SD 8.7; p=6.36×10−13) (table 1, online supplementary figure S8); they were also more likely to have ever smoked (44% vs 27%, p=0.004) and to be male (49% vs 37%, p=0.05). However, there was no difference in breathlessness between the PrePF and subjects without fibrosis (mean score 0.5 vs 0.6, p=0.24; table 3). The quantitative fibrosis score was positively associated with the breathlessness score (p=0.007), even after controlling for age (0.65), male sex (p=0.52) and smoking history (p=0.59). When fibrosis was defined by the quantitative fibrosis score cut-off (0.60), there was a trend towards higher breathlessness score in scans demonstrating lung fibrosis (0.44 vs 0.65, p=0.08).
Supplemental material
Dyspnoea questionnaire data
Screening for autoantibodies in this cohort revealed that there were no differences between PrePF and unaffected subjects in terms of overall seropositivity or specific antibody testing in this cohort (online supplementary table S4). For quantitatively defined lung fibrosis, there was also no significant difference between groups, with similar overall seropositivity rates (11% vs 16%, p=0.30).
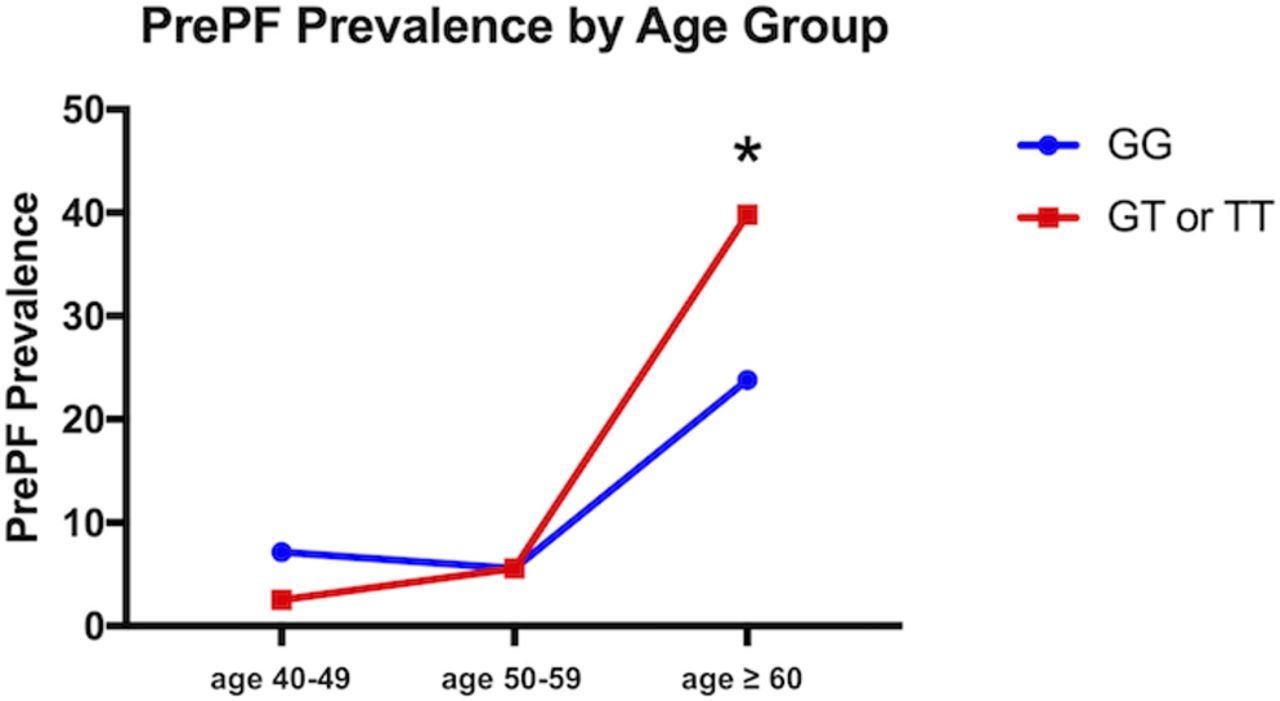
The MUC5B promoter variant rs35705950 was associated with the visual diagnosis of PrePF (present in 40% of those without fibrosis vs 53% with PrePF; MAF 0.21 vs 0.29, respectively, p=0.02; OR=2.14 (95% CI [1.00, 4.63], table 1). After age 60 years, there was a statistically significant difference in the proportion of subjects with visually diagnosed PrePF when the cohort was stratified by the MUC5B genotype (23.8% vs 39.8% prevalence, p=0.02); prior to age 60 years, PrePF prevalence is not significantly different by genotype (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prevalence of PrePF in FIP siblings cohort by age and MUC5B genotype. PrePF prevalence in this FIP siblings cohort increases by age, as shown in this graph. By age >60 years, the prevalence of PrePF differed significantly based on the MUC5B genotype (*p=0.2). Subjects with the variant are depicted by the red line, while those without it are depicted with the blue line. FIP, familial interstitial pneumonia; PrePF, preclinical pulmonary fibrosis.
MUC5B variant carriers, regardless of their visual CT diagnosis, had significantly higher qHRCT fibrosis scores (mean difference 0.36, p=0.006). The association between the MUC5B genotype and the fibrosis score was significant even when controlling for age, sex and smoking history in a linear regression (p=0.017, table 4). Age (p<2.0×10−16) was significantly associated with fibrosis score, but male sex (p=0.26) was not; the association of smoking and fibrosis score was borderline (p=0.05). The simpler quantitative scoring method, log %HAA, was not significantly different in MUC5B variant carriers (p=0.4).
Subject characteristics based on quantitative fibrosis score
When the 341 subjects with a visual inspection negative for fibrosis were separated further by whether or not qHRCT score indicated fibrosis, 59 were identified to have lung fibrosis by the deep learning method, and 282 were found to be unaffected. In those that were classified negative by both visual and computational methods, the mean age was 54.7 (95% CI 53.8 to 55.6); 101 (35.8%) were male; and 271 had genotyping available (MAF of 0.21 for the MUC5B promoter variant). Of those that were classified negative visually but fibrotic by deep learning (n=59), the mean age was 61.2 (95% CI 58.4 to 65.0); 22 were male (37.3%); and all had genotyping available, which revealed a MAF of 0.26 for the MUC5B promoter variant. Those that were identified as having lung fibrosis by deep learning were older (61.2 vs 54.7 years, p=4.2×10-5) and were more likely to have the MUC5B variant (MAF=0.26 vs 0.21, p=0.18); however, the MUC5B promoter variant association in this subanalysis did not reach statistical significance.
In contrast to the MUC5B variant, the common IPF-associated TERT variant (rs2736100) was not significantly associated with PrePF assessed either qualitatively (MAF 0.47 in PrePF vs 0.46 in unaffected, p=0.77) or quantitatively (MAF 0.50 fibrotic vs 0.47 not fibrotic, p=0.40).
When these factors were examined for their contributions to risk of PrePF in our study cohort, we used a mixed effects logistic regression model to test the independent effects of age sex, smoking and MUC5B or TERT genotypes while controlling for family. Age remained significantly associated with PrePF (OR 1.15, 95% CI 1.09 to 1.21, p=6.74×10−7), and the MUC5B variant was more common in PrePF (OR 2.14, 95% CI 1.00 to 4.63, p=0.05) (table 1). The common TERT variant (rs2736100) associated with fibrotic idiopathic interstitial pneumonia5 was not associated with PrePF in simple comparison of allele frequency (MAF was 0.45 in PrePF vs 0.45 in unaffected, p=0.92) or in a model controlling for age, sex, smoking history and family relatedness (p=0.38) (table 1).
Secondary subgroup analyses
Given the presence of non-fibrotic ILD (n=16, figure 1) in the ‘no fibrosis’ cohort, secondary analyses were performed that (1) excluded non-fibrotic ILDs (online supplementary table S5) and (2) compared all ILDs (inclusive of non-fibrotic ILD) with those without any ILD (online supplementary table S6). When non-fibrotic ILDs were excluded from the analyses, subjects with PrePF were older (p=1.7×10−12), more commonly male (p=0.05), more often had a smoking history (p=0.003) and had a higher prevalence of the MUC5B promoter variant (MAF 0.29 vs 0.20, p=0.02). However, when controlling for family relatedness and the other risk factors in a mixed effects logistic regression, age was associated with PrePF (OR 1.15, 95% CI 1.09 to 1.21, p=8.8×10−7), and the MUC5B promoter polymorphism had a borderline association with PreP (OR 2.15, 95% CI 0.99 to 4.69, p=0.05) (online supplementary table S5). Another secondary analysis of the data was performed in which all subjects with CT findings of ILD (fibrotic or non-fibrotic) were compared with those without any evidence of ILD (online supplementary table S6). Those with CT evidence of any ILD were older (mean age 64.5 years, SD 10.2) compared with those without any evidence of ILD (mean age 55.7 years, SD 8.7, p=7.2×10−12), more likely to be male (p=0.02), more likely to have smoked (p=0.0003) and more likely to carry the MUC5B promoter variant (MAF 0.29 vs 0.21, p=0.01). When controlling for family relatedness in a mixed effects logistic regression model, age (OR 1.11, 95% CI 1.07 to 1.15, p=5.58×10−9) and the MUC5B promoter variant (OR 1.87, 95% CI 1.04 to 3.36, p=0.04) were significantly associated with risk of ILD; smoking history had a borderline association (OR 1.81, 95% CI 1.01 to 3.25, p=0.05).
Discussion
ILAs have been studied previously in FIP relatives14; our present study builds on these initial findings by presenting data from a larger cohort, focusing specifically on evidence of fibrotic radiologic abnormalities and using qHRCT analysis. PrePF is common among first-degree relatives of patients with FIP, texture-based qHRCT analysis is useful in identifying these abnormalities in this population, and key factors predict those most at risk of this disease. PrePF subjects are older, more likely to be male and more likely to have smoked than the subjects without fibrosis.1 Additionally, the gain-of-function MUC5B promoter variant rs35705950, which is associated with established pulmonary fibrosis,4 5 7 25–29 is more common in PrePF subjects when compared with their unaffected family members. Given the high prevalence of findings suggestive of a UIP pattern on HRCT scan among subjects with PrePF and the association of IPF risk factors (age, gender, cigarette smoking and MUC5B promoter variant) with PrePF, our findings suggest that PrePF subjects are at risk of developing progressive fibrosis and that quantitative CT imaging represents a sensitive means of detecting these radiological abnormalities.
Even in a population such as first-degree relatives of patiens with FIP that is at baseline enriched for the MUC5B variant compared with the general NHW population,4 8 the MUC5B variant is more common in those with PrePF. A study of this variant in larger at-risk populations is necessary to determine if the genotype could be used to target prospective screening, especially in those over the age of 60 (figure 6). It is important to note, however, that the prevalence of PrePF was relatively high even in those without the MUC5B variant, suggesting that the absence of this variant alone may not indicate that a particular individual in this at-risk cohort would not warrant screening. Notably, we examined another IPF-associated common variant in TERT in this cohort and did not find that variant to be associated with PrePF; it is possible that due to the high MAF of the TERT variant in the general population, this study was underpowered to detect its relationship to risk of PrePF.
The deep learning method presented here is capable of detecting and quantifying fibrotic ILD patterns on CT in this cohort. Prior studies using a similar method17 used established IPF cases and correlated quantitative scores with pulmonary function testing, suggesting that a quantitative HRCT score reflects physiological change in addition to CT change. The current study supports the use of quantitative HRCT analysis to detect PrePF in a cohort of high-risk subjects without known disease since it is associated with breathlessness and the MUC5B promoter variant,4 5 27 a known IPF risk factor. However, the negative predictive value (97%) of using a quantitative fibrosis score cut-off was noted to be much higher than its positive predictive value (46%), suggesting that it may be particularly useful in terms of identifying higher risk scans that may require more careful visual inspection by radiologists. Recent studies illustrating that ILAs are under-reported in real-world settings suggest that technology-aided evaluation of routine chest imaging could improve timeliness of patient referral and evaluation.30
This deep learning method based on textural analysis appears to be superior to %HAA, a simpler densitometry-based method that has been applied to quantitative CT assessment of ILDs.24 Compared with the deep learning fibrosis score, the %HAA method was less accurate, had a lower positive predictive value and was not associated with the MUC5B risk variant. While the %HAA method of HRCT analysis may capture some forms of PrePF, more advanced methods of quantifying subtle fibrosis are needed to quantify these features consistently. A limitation of deep learning is the need for a significant amount of labelled training data. The CNN used for the present study was trained using an independent dataset composed of subjects enrolled in a clinical trial for IPF.17 These subjects had more advanced lung fibrosis than those in this cohort, and their HRCT technical parameters were more consistent.
Though this study was performed on a cohort of first-degree relatives of patients with FIP, we hypothesise that the findings could be relevant to first-degree relatives of patients with sporadic IPF. Given that genome-wide studies have shown that FIP and sporadic IPF are indistinguishable in terms of common risk variants,5 a hypothesis that should be tested is that the genetic and genomic markers identified through the study of PrePF in FIP families could be applicable to first-degree relatives of patients with sporadic IPF. Additional genetic variants, both common and rare, associated with fibrotic ILD could be examined in this cohort to determine how they contribute to risk of PrePF. Due to lack of power especially for common variants, larger cohorts would be required to determine additive effects of multiple genetic variants in terms of PrePF risk.
We hypothesise that PrePF could represent an early form of IPF. Prior studies illustrate that ILAs are associated with progressive loss of lung function and increased mortality,12 13 suggesting that the abnormalities we observe here, like ILAs studied in other cohorts,8 31 32 may have clinical consequences. However, longitudinal observation of these subjects is required to determine at what rate and with what frequency PrePF progresses among FIP relatives in particular and whether it behaves like IPF. In addition, our ability to determine whether PrePF progresses to clinical IPF (vs other progressive fibrotic lung diseases) is limited by the fact that we do not have verified data regarding the subjects’ previous environmental and occupational exposures—a clinical diagnosis of IPF would necessitate exclusion of environmental or occupational exposures associated with fibrosis with extensive interviewing. Given the differing age distributions of those with and without PrePF, it is likely that a substantial proportion of study subjects that had HRCTs without evidence of fibrosis at this one point in time may develop pulmonary fibrosis as they age. Further characterisation of PrePF is necessary before we can determine how these findings should be applied to counselling and potential screening of relatives of patients with FIP.
Currently, the MUC5B promoter variant and quantitative methods of CT analysis are not used to assist in the clinical detection of PrePF. Future studies will further phenotype PrePF in this population. Other genetic variants (rare and common) associated with pulmonary fibrosis will be examined to determine the relative importance of different risk alleles in this population. Longitudinal study is required to determine the ability of the deep learning method of HRCT analysis to detect parenchymal changes that may precede fibrosis identified by standard visual examination.
In conclusion, PrePF is common in relatives of patients with FIP and associated with age, as well as the MUC5B promoter variant. Quantitative HRCT scoring using deep learning is capable of detecting PrePF and is associated with the MUC5B promoter variant, breathlessness symptoms, and visual diagnosis of fibrotic ILD.
Supplemental material
Supplemental material
Supplemental material
References
Footnotes
Contributors SKM compiled and analysed visual CT reads, performed genotyping, statistical analyses, wrote first draft of manuscript and revised the manuscript in collaboration with the other authors. SH created and implemented the quantitative high-resolution CT algorithm described in this article; he also performed %HAA quantification on CT scans in this study and contributed to the first draft of the manuscript. JP led research coordination, data management and study subject recruitment at the University of Colorado. ADW performed nucleic acid extractions and sample management for this project. JW contributed to data management and study subject recruitment. IVY contributed to and advised on the overall study design, genetic analyses and gene expression analyses. TF oversaw statistical analyses presented in this article. KKB and MPS were integral to study subject recruitment. MIS contributed to the overall study design. DAL and JHC. performed radiological reviews for this study. CM, JAK, TSB and JEL led patient recruitment, study design, and data and sample management at Vanderbilt University. DAS led overall study design and subject recruitment, and contributed to each stage of data analyses and manuscript drafting. All authors contributed to manuscript revisions.
Funding NIH-NHLBI (UH2/3-HL123442, R01-HL097163, R21/R33-HL120770, P01-HL092870, K23-HL136785, K08-HL130595, F32HL123240), U.S. DOD (W81XWH-17-1-0597).
Competing interests DAS is the founder and chief scientific officer of Eleven P15, a company focused on the early diagnosis and treatment of pulmonary fibrosis. DAS has an awarded patent (US patent no: 8,673,565) for the treatment and diagnosis of fibrotic lung disease. DAL and SMH have a pending patent (application US20170330320A1) for image analysis; SMH reports a consulting agreement with Boehringer Ingelheim.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Anonymized data are available upon reasonable request.