Article Text

Abstract
Introduction Dysregulated sphingolipid metabolism has been implicated in the pathogenesis of various pulmonary disorders. Nuclear sphingosine-1-phosphate (S1P) has been shown to regulate histone acetylation, and therefore could mediate pro-inflammatory genes expression.
Methods Profile of sphingolipid species in bronchoalveolar lavage fluids and lung tissue of mice challenged with Pseudomonas aeruginosa (PA) was investigated. The role of nuclear sphingosine kinase (SPHK)2 and S1P in lung inflammatory injury by PA using genetically engineered mice was determined.
Results Genetic deletion of Sphk2, but not Sphk1, in mice conferred protection from PA-mediated lung inflammation. PA infection stimulated phosphorylation of SPHK2 and its localisation in epithelial cell nucleus, which was mediated by protein kinase C (PKC) δ. Inhibition of PKC δ or SPHK2 activity reduced PA-mediated acetylation of histone H3 and H4, which was necessary for the secretion of pro-inflammatory cytokines, interleukin-6 and tumour necrosis factor-α. The clinical significance of the findings is supported by enhanced nuclear localisation of p-SPHK2 in the epithelium of lung specimens from patients with cystic fibrosis (CF).
Conclusions Our studies define a critical role for nuclear SPHK2/S1P signalling in epigenetic regulation of bacterial-mediated inflammatory lung injury. Targeting SPHK2 may represent a potential strategy to reduce lung inflammatory pulmonary disorders such as pneumonia and CF.
- bacterial infection
- respiratory infection
- airway epithelium
- pneumonia
- cystic fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
Does nuclear sphingosine-1-phosphate (S1P) signalling regulates bacteria-induced lung inflammation?
What is the bottom line?
Elevated nuclear sphingosine kinase 2 (SPHK2) activity following Pseudomonas aeruginosa (PA) infection of the lung resulted in increased nuclear S1P production that epigenetically regulated the secretion of pro-inflammatory cytokines.
Why read on?
We, for the first time, report that PA infection of lung epithelium stimulates nuclear localisation of SPHK2 mediated by protein kinase C δ that results in enhanced production of S1P in the nucleus.
We further demonstrate that PA-mediated secretion of pro-inflammatory cytokines is epigenetically regulated by nuclear S1P via histone deacetylases and H3 and H4 histone acetylation of interleukin-6 promoter. Furthermore, genetic deletion of Sphk2 in mice or inhibition of SPHK2 activity postinfection mitigated PA-induced lung inflammatory injury lending support to the concept that SPHK2 may be a therapeutic target.
Introduction
Pseudomonas aeruginosa (PA), a Gram-negative opportunistic pathogen, causes clinical pneumonia in humans, and is associated with significant morbidity and mortality. PA-mediated pulmonary infection is prevalent in people with cystic fibrosis (CF),1 2 chronic obstructive pulmonary disease (COPD) 3 4 and in those on mechanical ventilation.5 Clinical features of PA-induced acute inflammation are evidenced by rapid (min to hour) infiltration of neutrophils as the major cellular infiltrate and mildself-limiting tissue injury; however, acute infection in certain populations, such as ventilator-associated pneumonia and acute pneumonia in neutropenic patients can be severe and not self-limiting. On the contrary, chronic inflammation is slow (days) with monocytes/macrophages/lymphocytes as the major infiltrating cells and the tissue injury is severe and progressive.6 Interestingly, PA rarely infects normal human lungs without an underlying defect in immunity or a breach in the mechanical barrier as the clearance of the bacteria is rather efficient. The pathogen is recognised by membrane-bound pattern recognition receptors, known as toll-like receptors (TLRs) expressed on the host epithelium. Epithelial cells that line the respiratory tract and small airways are crucial site for innate immune responses, and PA expresses numerous pathogen-associated molecular patterns such as lipopolysaccharide (LPS) and flagellin. Based on the studies conducted in a murine model of PA-induced pneumonia, it was shown that the host sensing of LPS is mostly by TLR47 and possibly TLR28 while flagellin is recognised by TLR5.9
PA infection of the lung activates host innate immune responses,10 increases reactive oxygen species (ROS) generation via NADPH oxidase (NOX) proteins11 and modulates sphingolipid metabolic pathways.12 Among the sphingolipids, ceramides generated from sphingomyelin by acid sphingomyelinase activation are known to accumulate in the airway epithelium of CF patients with Pseudomonas infection. In contrast, sphingosine, generated from ceramides by ceramidases, is present in healthy airways and almost entirely absent in Pseudomonas-infected CF lungs.12 It has been postulated that high concentration of ceramide in PA-infected lungs promotes adhesion, invasion and migration while high levels of sphingosine in healthy cells kills invading bacteria.13 Although signalling via sphingolipids, in particular ceramide, has been implicated in host-bacterial interactions, the mechanism(s) underlying the involvement of sphingolipids in PA-induced lung infection and subsequent clearance remains to be established.
Sphingomyelin is the major sphingolipid present in biological membranes and is synthesised de novo from serine and palmitoyl coenzyme A by serine palmitoyl transferase to 3-keto-dihydrosphigosine, which is subsequently reduced to dihydrosphingosine and converted to ceramide.14 Ceramide serves an intermediate for sphingomyelin biosynthesis or hydrolysed to sphingosine by ceramidase.15 Sphingosine-1-phosphate (S1P) is biosynthesised in mammalian cells by phosphorylation of sphingosine catalysed by sphingosine kinase (SPHK)1 and 2 and catabolised by S1P phosphatases and S1P lyase.16 SPHK2 with a functional nuclear localisation signal is a nuclear protein,17 while SPHK1 is a cytosolic enzyme.18 We and others have demonstrated that the bioactive sphingolipid, S1P signalling via the G-protein coupled S1P1-5 contributes to the development of several lung pathologies such as sepsis, pulmonary arterial hypertension (PAH),19 asthma,20 pulmonary fibrosis (PF)21 22 and bronchopulmonary dysplasia (BPD).23 Genetic deletion of Sphk1, but not Sphk2, in mouse potentiated LPS-induced lung inflammation and pulmonary oedema suggesting a protective role of SPHK1/S1P signalling axis in experimental sepsis.24 Furthermore, partial deletion or inhibition of S1P lyase conferred protection against LPS-induced lung injury in mice.25 In contrast to sepsis, genetic deletion of Sphk1 or inhibition of SPHK1 ameliorated lung inflammatory injury and pulmonary oedema in lung pathologies such as asthma,26 PF,21 PAH19 and BPD.23 S1P generation in murine skin increases cathelicidin antimicrobial peptide that regulates epithelial innate immune responses27 and plethora of studies have pointed to the role of S1PR signalling in trafficking, differentiation and activation of immune cell effector functions.28 However, very little is known on the role and modulation of sphingolipids in lungs after bacterial infection.
Our initial analysis of CF lung specimens revealed increased nuclear staining of p-SPHK2 in alveolar and bronchial epithelial cells compared with normal lungs. As chronic PA lung infection is a hallmark of CF, we sought to gain further insights into the role of dysregulated sphingolipid metabolism and signalling in PA-induced inflammatory lung injury in vivo and in vitro. In this study, we observed that genetic deletion of Sphk2, not Sphk1, or inhibition of SPHK2 activity in mice reduced PA-induced lung inflammation as evidenced by decreased levels of inflammatory cytokines such as interleukin (IL)-6 and tumour necrosis factor (TNF)-α. Moreover, PA infection of lungs in vivo and epithelial cells in vitro increased histone H3 and H4 acetylation, which was dependent on protein kinase C (PKC) δ-mediated nuclear SPHK2 phosphorylation and S1P production.
Materials and methods
For detailed materials and methods, see the online supplementary data.
Supplementary file 1
Human cystic fibrosis lung specimens
Six cases of advanced CF subjected to lung explantation were selected from the archives from the Department of Pathology of the Colorado Children’s Hospital. The CF lung donors were 4 males and 3 females, aged 16–24 years. These lungs had characteristic gross and microscopic features of CF. These consist of bronchiectasis and bronchiolectasis, with extensive peri-airway fibrosis. Microscopically, the airways had characteristic mucus accumulation admixed with large numbers of neutrophils. The inflammatory process extended to adjacent alveolar structures. Six normal lungs, not used for transplantation, were obtained from anonymous lung donors. They were histologically normal.
Results
Pseudomonas aeruginosa infection alters sphingolipid levels in mouse lung and bronchoalveolar lavage fluid
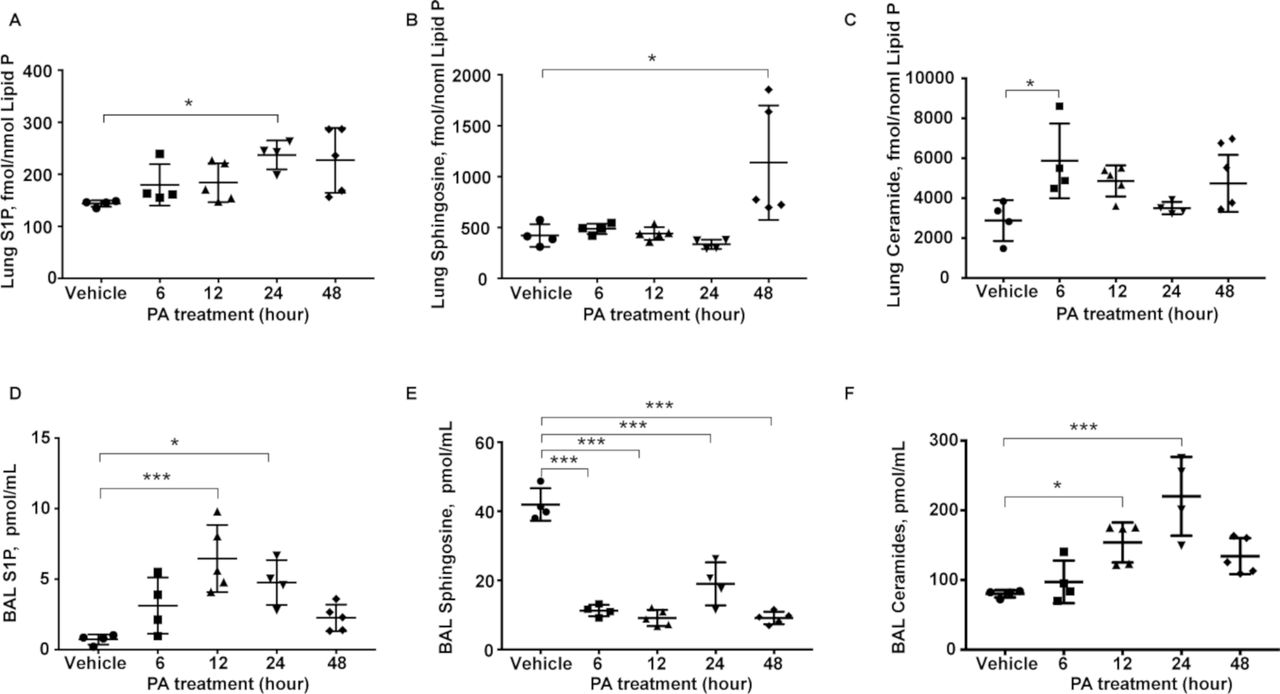
Earlier studies have shown that PA infection enhanced accumulation of ceramide and reduction of sphingosine in the airway12; however, not much is known on the role of other sphingoid bases in the lungs after bacterial infection. PA infection of mouse lung for 48 hours significantly altered sphingolipid levels, as quantified by mass spectrometry, in lung tissues and bronchoalveolar lavage (BAL) fluid. Ceramide and S1P levels were elevated in lung tissues as early as 6 hours postinfection, while sphingosine levels almost doubled at 48 hours (figure 1A-C). Similarly, S1P and ceramide levels in the BAL fluid were higher from 12 to 48 hours after PA infection; however, sphingosine levels were significantly lower in BAL fluid at all the time points (figure 1D-F). No significant changes in the expression of SPHK1, SPHK2 and S1P lyase were observed in total lung tissue lysates from control and PA-treated animals (data not shown). These results suggested that sphingoid base levels were modulated in mouse lung and BAL fluid after PA infection, which may play a role in the development of PA-mediated lung inflammation and injury, and generation of S1P in the lung. Furthermore, elevated sphingosine levels in the PA-infected lung at 48 hours may serve as a substrate for SPHKs to generate S1P.

PA infection alters sphingolipid levels in mouse lungs and BAL fluids. Wild-type mice were challenged intratracheally with either sterile PBS or PA 103 (1×106 CFU/animal) in a total volume of 50 µL for 24 hours. Animals were sacrificed, BAL fluid was collected, centrifuged and analysed. Lungs were removed and frozen in liquid N2 immediately. Lipids were extracted from BAL fluid and lung tissues. Quantification of S1P, sphingosine and ceramide levels in lung tissues and BAL fluids from control and PA103 infected mice (1×106 CFU/mouse) 24 hours postinfection by LC-MS/MS. (A–C) Sphingolipid levels in lung tissues (A) S1P, (B) sphingosine and (C) ceramide. (D–F) Sphingolipid levels in BAL fluid. (D) S1P, (E) sphingosine and (F) ceramide. n=5. Data are from one experiment with C57BL/6J mice (n=5; male and female) used for each treatment, and data are expressed as means±SE; *P<0.05; ***p<0.001. BAL, bronchoalveolar lavage; CFU, colony-forming unit; LC-MS/MS, liquid chromatography tandem-mass spectrometry; PA, Pseudomonas aeruginosa; PBS, phosphate buffered saline; S1P, sphingosine-1-phosphate.
Sphk2, but not Sphk1, deficient mice are protected from PA-mediated lung inflammation
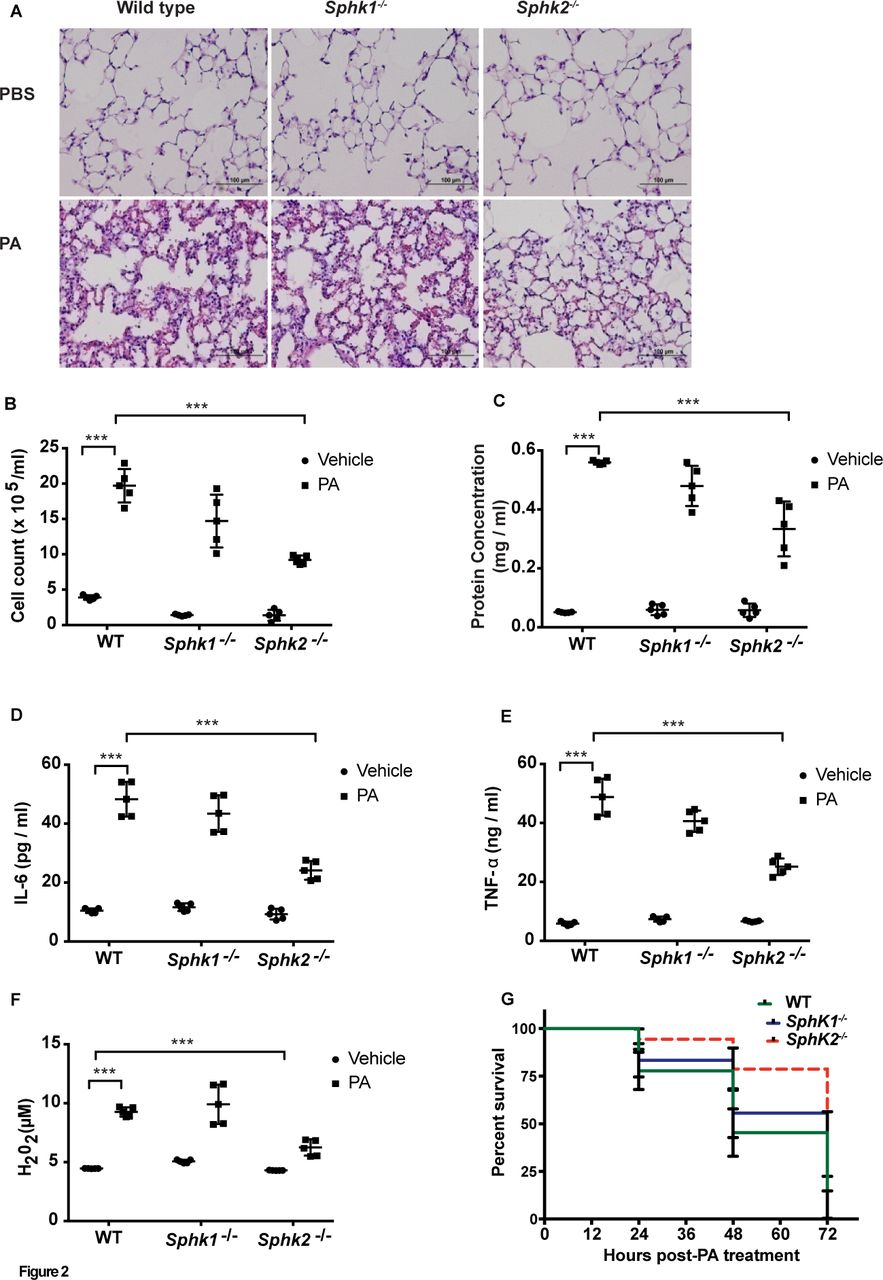
As tissue S1P levels are in part regulated by its synthesis and catabolism, we assessed the role of SPHK1 or SPHK2 in PA-mediated lung inflammation. Intratracheal instillation of PA (1×106 CFU/animal) induced lung injury, Peripheral blood mononuclear cells (PMN)infiltration in lungs and increased protein levels in BAL fluid in wild-type (WT) mice, whereas these responses were significantly lower in Sphk2-/- mice. In contrast, Sphk1 deletion in mice had no significant effect on PA-induced lung injury (figure 2A-C). In response to PA infection, concentrations of the pro-inflammatory mediators IL-6 and TNF-α and H2O2 were significantly elevated in WT mice (figure 2D-F), while IL-β and Macrophage Inflammatory Protein 2 (MIP2-α) showed a moderate change (online supplementary figure 1C and D).Genetic deletion of Sphk2, but not Sphk1, in mice significantly reduced neutrophil infiltration into the lung (online supplementary figure 1A and B) compared with the WT mice; however, Sphk2 deletion had no effect on PA-mediated IL-1β and MIP2 secretion (online supplementary figure 1C and D).In survival studies, ~50% of the WT and Sphk1-/- mice died at 48 hours postintratracheal instillation of PA at a higher dosage (1×107 CFU/animal), whereas ~75% of Sphk2-/- survived during the same time periods (figure 2G). No differences in BAL colony counts were seen between the WT, Sphk1-/- and Sphk2-/- mice (online supplementary figure 1E). These results suggested that deletion of Sphk2, but not Sphk1, protected mice against PA-mediated inflammatory lung injury.
Supplementary file 2

Deletion of Sphk2 prevents PA-induced lung inflammatory injury. C57BL/6J WT, Sphk1-/- and Sphk2-/- mice (same background) (n=5) were challenged intratracheally with sterile PBS or PA103 (1×106 CFU/mouse) for 24 hours. Lungs were removed, embedded in paraffin and cut into 5 µm sections for staining. (A) Representative H&E staining photomicrographs of lung sections from WT, Sphk1-/- and Sphk2-/- mice with/without PA challenge, original magnification, 10x, scale bar=100 µm. (B–F) BAL fluids from control and PA challenged WT, Sphk1-/- and Sphk2-/- mice were analysed for infiltrating cells, protein, pro-inflammatory cytokines and H2O2 levels. (B) Total infiltrated cells in BAL, (C) total protein levels in BAL fluid, (D) concentration of IL-6 in BAL fluid, (E) concentration of TNF-α in BAL fluid and (F) concentration of H2O2 in BAL fluids. Data are expressed as means±SE from one experiment (number of animals per group=5). ***P<0.001. (G) Survival of WT, Sphk1-/- and Sphk2 -/- mice exposed to high dose of PA. WT, Sphk1-/- and Sphk2-/- mice were challenged intratracheally either with sterile PBS or PA 103 (1×107 CFU/animal) in a total volume of 50 µL and animals were monitored for a period of 96 hours (n=10 in each group). Mice deficient in Sphk2 showed significantly lower mortality (~25%) compared with WT and Sphk1 KO mice. Significance was calculated using two-way analysis of variance. BAL, bronchoalveolar lavage; H2O2, hydrogen peroxide; IL-6, interleukin-6; PA, Pseudomonas aeruginosa; PBS, phosphate buffered saline; SphK1, sphingosine kinase 1; SphK2, sphingosine kinase 2; TNF-α, tumour necrosis factor α; WT, wild type.
Inhibition of SPHK2 activity reduced PA-induced lung inflammation
To further examine the potential role of SPHK2 activity in lung inflammatory injury after PA challenge, we evaluated the effect of ABC294640, a specific inhibitor of SPHK2.29 When compared with vehicle-challenged mice, treatment with ABC294640 (10 mg/kg body weight) 1 hour prior to intratracheal PA exposure for 24 hours reduced infiltration of inflammatory cells into alveolar space and lung inflammation and injury (online supplementary figure 2A-D). Furthermore, PA-mediated lung injury, infiltration of inflammatory cells in the lungs, increased levels of pro-inflammatory cytokines and H2O2 levels in BAL fluid were significantly reduced in WT mice treated with ABC294640 6 hours postinfection compared with vehicle challenged mice (figure 3A-F). To determine the off-target effect(s) of the SPHK2 inhibitor, WT and Sphk2-/- mice were administered ABC294640 (10 mg/kg body weight, intraperitoneal) 1 hour prior to PA challenge and sphingoid base levels and infiltration of cells were determined. As shown in online supplementary figure 3A, S1P levels were increased in BAL fluid from WT and Sphk2-/- mice challenged with ABC294640; however, ABC294640 had no significant effect on sphingosine and ceramide levels in BAL fluid from WT and Sphk2 KO mice (data not shown). Interestingly, genetic deletion of Sphk2 in mice also enhanced S1P levels in BAL fluid of control and PA-infected animals (online supplementary figure 3A). These results are in agreement with earlier reports demonstrating that inhibition of SPHK2 in mice increases S1P levels in plasma and tissues.30–32 Furthermore, administration of ABC294640 had no significant effect on infiltration of inflammatory cells, protein and pro-inflammatory cytokine levels in BAL fluid (online supplementary figure 3B-E). Taken together, these results suggested that inhibition of SPHK2 postinfection may have a therapeutic beneficiary effect in reducing lung inflammatory injury and ruled out off-target effect(s) of ABC294640 on PA-mediated lung inflammation.

Inhibition of SPHK2 with ABC 294640 ameliorates PA-induced inflammatory lung injury. WT C57BL/6J mice were challenged intratracheally with sterile PBS (n=10) or PA103 (n=10) as outlined in figure 1. After 6 hours of PA instillation, five mice from each group received the SPHK2 inhibitor ABC 294640 (10 mg/kg body weight), dissolved in DMSO, intraperitoneally; five mice from control group also received equal volume of DMSO (1 µL in 100 µL). All animals were sacrificed at the end of 24 hours postinfection of PA. Animals were sacrificed; BAL fluid was collected, and lungs were removed, embedded in paraffin and cut into 5 µm sections for staining. (A) Representative H&E staining photomicrographs of lung sections from control, PA, ABC294640 and ABC294640+PA-treated animals. Original magnification, 10x, scale bar=200 µm. (B) Total infiltrated cells in BAL fluid, (C) total protein levels in BAL fluids, (D) concentration of IL-6 in BAL fluids, (E) concentration of TNF-α in BAL fluids and (F) concentration of H2O2 in BAL fluids. Data are expressed as means±SE from one experiment (number of animals per group=5). *P<0.05; **p<0.01; ***p<0.001. BAL, bronchoalveolar lavage; DMSO, dimethyl sulfoxide; PA, Pseudomonas aeruginosa; IL-6, interleukin 6; PBS, phosphate buffered saline; TNF-α, tumour necrosis factor α.
PA stimulated SPHK2 phosphorylation and nuclear localisation in lung epithelium in vivo and in vitro
To understand the role of SPHK2 in regulating inflammation, we next determined if SPHK2 activation is regulated by post-translational modification such as phosphorylation. Infection of mouse lungs with PA enhanced localisation of p-SPHK2 in the nucleus of alveolar type II (ATII) epithelial cells (figure 4A). Total homogenates from PA-infected lungs revealed enhanced phosphorylation of SPHK2 (figure 4B), and quantification of p-SPHK2 staining of paraffin-embedded lung tissues revealed significantly higher proportion of nuclear p-SPHK2 staining in ATII cells compared with human bronchial epithelial cells (HBEpCs) (figure 4C & D).
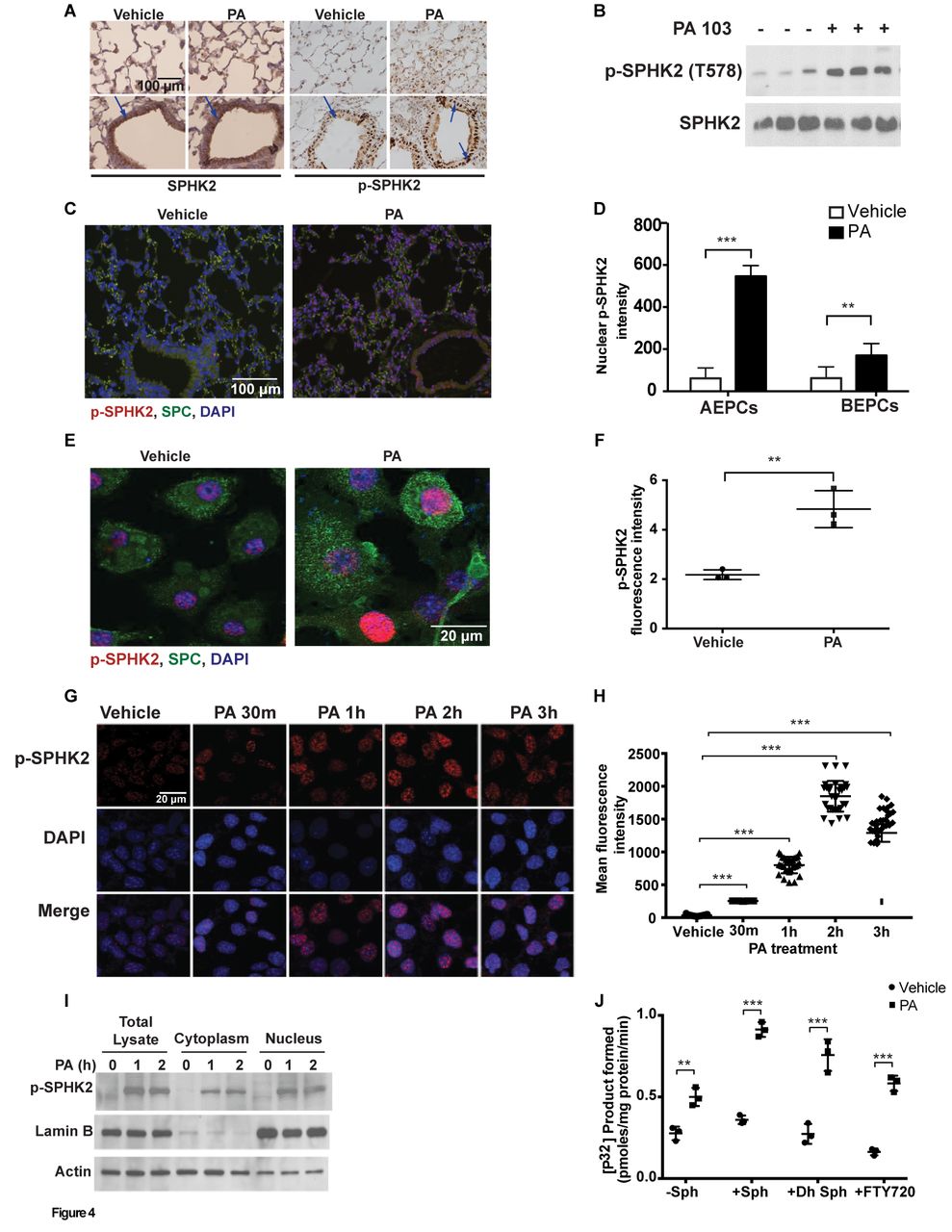
PA stimulates SPHK2 phosphorylation and nuclear localisation in lung epithelium. (A) Paraffin-embedded lung tissues from WT and PA-infected mice were immunostained with SPHK2 and p-SPHK2 antibodies, original magnification, 10x, scale bar=100 µm. (B) Lung tissue lysates from vehicle and PA103 instilled animals were analysed for SPHK2 and p-SPHK2 by immunoblotting. Shown is representative blot from five independent experiments. (C) Co-immunostaining of p-SPHK2 and epithelial marker, SPC in lung specimens from WT control and PA-challenged mice, original magnification, 10x, scale bar=100 µm and (D) quantification of the co-immunostaining of p-SPHK2 and SPC in ATII and HBEpCs. (E) Primary ATII cells isolated from control and PA challenged mouse lung were co-immunostained for p-SPHK2 and SPC, original magnification, 60x, scale bar=20 µm. Shown is a representative co-immunostained image (five areas per slide; four slides per group). (F) Quantification of (E) by ImageJ. (G and H) MLE-12 cells grown on slide chambers were challenged with vehicle or heat-inactivated PA103 (1×108 CFU/mL) for varying time periods, immunostained for p-SPHK2 and nucleus by DAPI. (G) Shown is a representative image from four independent experiments, original magnification, 60x, scale bar=20 µm. (H) Quantification of co-stained images of p-SPHK2 and DAPI was performed (five areas per slide; four slides per group) using ImageJ software. (I) Western blot analysis of total lysate, cytoplasm and nuclear fractions isolated from control and PA103 challenged MLE-12 cells for 0, 1 and 2 hour. (J) MLE-12 cells in 100 mm dishes were treated with vehicle or heat-inactivated PA for 2 hours followed by isolation of nuclear fraction and measurement of SPHK activity. **P<0.01; ***p<0.001. ATII, alveolar type II; DAPI, 4’,6-diamidino-2-phenylindole; HBEpCs, human bronchial epithelial cells; MLE-12, murine lung epithelial-12; PA, Pseudomonas aeruginosa; PBS, phosphate buffered saline; SPHK, sphingosine kinase; SPHK2, sphingosine kinase 2; SPC, surfactant protein C; WT, wild type.
We next investigated whether phosphorylation and nuclear localisation of p-SPHK2 occurred in vitro in lung epithelial cells. Exposure of primary ATII cells (figure 4E & F), mouse alveolar epithelial (MLE-12) cell line (figure 4G & H) or primary HBEpCs (online supplementary figure 4A and B) to heat-inactivated PA for 3 hours stimulated nuclear localisation of p-SPHK2 as determined by immunofluorescence, which was verified by isolation of nuclear fractions from MLE-12 cells. As shown in figure 4I, stimulation of MLE-12 cells with heat-inactivated PA enhanced p-SPHK2 localisation in the nucleus and the purity of the nuclear fraction was verified by lamin B1, a marker for the nucleus. Activation of SPHK2 in the nucleus of MLE-12 cells increased nuclear S1P levels (online supplementary figure 5C) while the levels of other sphingoid bases were not significantly altered both in the cytoplasm and nuclear fractions (online supplementary figure 5A and B, D-F). The nuclear fractions from PA challenged MLE-12 cells showed increased phosphorylation of sphingosine, dihydrosphingosine and FTY720 to the corresponding sphingoid phosphates as compared with controls (figure 4J). These results showed that PA infection stimulated SPHK2 phosphorylation, its nuclear localisation and elevated nuclear S1P levels in lung epithelial cells.
Deletion or inhibition of SPHK2 reduced PA-mediated H3 and H4 histone acetylation and IL-6 secretion
Increased acetylation of histone protein H3 on the promoters of pro-inflammatory genes plays a key role in cigarette smoke33 and lipopolysaccharide-induced lung inflammation34; however, signalling mechanisms that regulate chromatin remodelling have not been extensively studied in bacterial infection. We next investigated whether PA modulated chromatin remodelling in vivo and in vitro. Infection of mouse lungs with PA increased H3K9 and H4K8 histone acetylation, which was reduced in Sphk2, but not Sphk1, knockout mice (figure 5A), and blocking SPHK2 activity in mouse lungs with ABC294640 attenuated H3K9 and H4K8 histone acetylation without affecting the total expression of H3 and H4 histones (figure 5B). In vitro, pretreatment of MLE-12 cells with ABC294640 attenuated H3K9 and H4K8 histone acetylation (figure 5C); however, inhibition of SphK1 activity with a specific inhibitor, PF-543, had no significant effect on H3 and H4 histone acetylation (figure 5C). Additionally, both ATII epithelial cells and HBEpCs exhibited enhanced H3K9 and H4K8 histone acetylation after treatment with PA (online supplementary figure 4C), and downregulation of Sphk2 with a specific siRNA in HBEpCs attenuated PA-mediated H3 and H4 histone acetylation in HBEpCs (online supplementary figure 4D). As levels of acetylated H3 and H4 histones are regulated by histone acetyl transferases (HATs) and histone deacetylases (HDACs), we next measured HDAC activity in epithelial cells after PA challenge. As shown in figure 5D, PA challenge inhibited total HDAC activity in ATII epithelial cell lysates, which could account in part for the enhanced H3 and H4 acetylation. Inhibition of SPHK2 activity with ABC294640 restored PA-dependent increase in nuclear HDAC activity but not in the cytoplasmic fraction isolated from MLE-12 cells (online supplementary figure 4E and F).
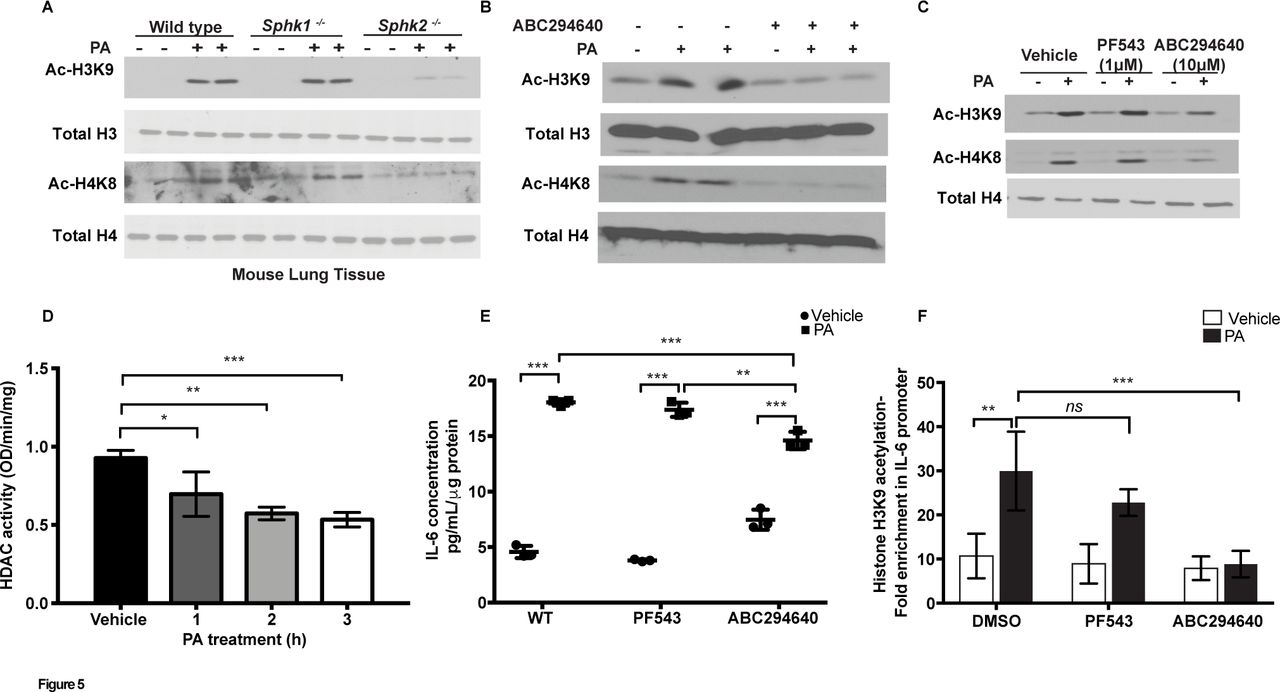
SPHK2 is essential for PA-induced H3 and H4 histone acetylation and IL-6 secretion. (A) WT C57BL/6J, Sphk1-/- and Sphk2-/- mice were challenged intratracheally with sterile PBS or PA103 as outlined in figure 2. Lung tissue lysates (30 µg proteins) were subjected to immunoblot analysis using antibodies to acetylated and total histones. Shown is a representative western blot analysis from five lung tissue lysates. (B) Lung tissue lysates (30 µg proteins) from control, PA, ABC294640, ABC294640+PA challenged mice, as described in figure 3, were analysed for histone acetylation. Shown is a representative blot from five lung tissue lysates subjected to western blot analysis. (C) MLE-12 cells were pretreated with vehicle or the SPHK1 inhibitor, PF-543 (1 µM) or SPHK2 inhibitor, ABC294640 (10 µM) for 1 hour prior to challenge with heat-inactivated PA (1×108 CFU/mL) for 2 hours. Cell lysates (20 µg proteins) were subjected to immunoblotting and immunostaining with acetylated histone antibodies. Shown is a representative blot of three independent experiments in triplicate. (D) Primary ATII cells were treated with heat-inactivated PA (1×108 CFU/mL) for 1–3 hours and cell lysates were analysed for HDAC activity using a commercial kit as outlined in ’Materials and methods' section. Values are means±SE of three experiments in triplicate and normalised to total protein per min. (E) Media from (C) were analysed for IL-6 levels using a commercial ELISA as described in ’Materials and methods' section. (F) ChIP assays were performed using antiacetyl histone H3K9 followed by real-time quantitative PCR. Data are means±SE from three independent experiments in triplicates. *P<0.05, **p<0.01; ***p<0.001. ATII, alveolar type II; ChIP, chromatin immunoprecipitation; HDAC, histone deacetylase; IL, interleukin; MLE-12, murine lung epithelial-12; ns, not significant; PA, Pseudomonas aeruginosa; PBS, phosphate buffered saline; SPHK1, sphingosine kinase 1; SPHK2, sphingosine kinase 2; WT, wild type.
Having demonstrated a role for SPHK2 in PA-induced H3 and H4 histone acetylation, we next examined the role of SPHK1 and SPHK2 on IL-6 secretion. In MLE-12 cells, PA stimulated IL-6 secretion (~threefold) was attenuated by inhibition of SPHK2, but not SPHK1, activity compared with control cells not treated with the inhibitors (figure 5E). To further examine whether PA can modulate histone acetylation pattern at IL-6 promoter, chromatin immunoprecipitation (ChIP) assays were performed using antiacetyl histone H3K9 antibody. PA significantly increased H3K9 histone acetylation at nuclear factor kappa B (NF-κB) binding sites on IL-6 promoter at 3 hour post-infection, which was blocked by SPHK2 inhibitor ABC294640 and not by SPHK1 inhibitor PF-543 (figure 5F). Collectively, these results showed for the first time a role for SPHK2 activation in PA-mediated H3 and H4 acetylation in vivo and in vitro, and regulation of chromatin modification in IL-6 promoter regions of NF-κB binding site(s) by PA-mediated SPHK2 activation.
Activation of PKC δ is essential for PA-induced SPHK2 phosphorylation, H3 and H4 histone acetylation and IL-6 secretion
Earlier studies have demonstrated a role for ERK1/2 in agonist-mediated phosphorylation and activation of SPHK1 and SPHK2 in mammalian cells.35 36 PA challenge of MLE-12 cells stimulated ERK1/2 phosphorylation; however, inhibition of ERK1/2 phosphorylation with PD98059 or U0126 (inhibitors of MEK1/2) had no effect on PA-mediated IL-6 secretion (data not shown). As exogenous S1P is an activator of PKC,37 we determined the role of PKC isoforms in PA-mediated SPHK2 activation and lung inflammation. Mouse and human alveolar and airway epithelial cells expressed most of the PKC isoforms,38 and PA challenge of MLE-12 cells enhanced total PKC activity in a time-dependent manner (figure 6A). Pretreatment of cells with a pan PKC inhibitor, bisindolylmaleimide blocked PA-induced SPHK2 phosphorylation and histone acetylation (figure 6B). To determine the role of PKC isoform(s), MLE-12 cells were infected with dominant negative (DN) PKC α, δ, ε and ζ isoforms prior to PA challenge. Among the PKC isoforms, DN Pkc α had no effect on IL-6 secretion while DN PKC δ, ε and ζ attenuated PA-induced IL-6 secretion (online supplementary figure 6A). Furthermore, downregulation of PKC δ expression with siRNA reduced SPHK2 phosphorylation and acetylation of H3K9 and H4K8 in MLE-12 cells challenged with PA as compared with control cells (figure 6C), and overexpression of DN Pkc δ reduced PA-induced il-6 and Tnf-α mRNA expression (online supplementary figure 6B and C) and IL-6 secretion (figure 7A). Also, DN Pkc δ, in a time-dependent manner, significantly attenuated PA mediated p-SPHK2 immunostaining in the nucleus as compared with the vector control (figure 6D & E). Next, ChIP assays were performed to determine if inhibition of PKC δ would modulate PA-mediated H3K9 histone acetylation at NF-κB binding sites on IL-6 promoter. Real-time quantitative PCR analysis showed that PA significantly increased H3K9 histone acetylation at NF-κB binding sites on IL-6 promoter, which was blocked by overexpression of DN PKC δ in MLE-12 cells (figure 7B). Furthermore, pretreatment of MLE-12 cells with SPHK2 inhibitor ABC294640 or adenoviral DN Pkc δ attenuated PA-induced increase in nuclear S1P levels (figure 7C). These results showed that PA-induced activation of PKC δ stimulated SPHK2 phosphorylation, H3 and H4 histone acetylation and secretion of IL-6 in lung epithelial cells.

PKC δ activation regulates PA-induced SPHK2 phosphorylation and histone acetylation. (A) MLE-12 cells were exposed to heat-inactivated PA (1×108 CFU/mL) for 1–3 hours and total PKC activity in cell lysates was determined using a commercial PKC activity kit. Values are means±SE from three independent experiments in triplicate. **P<0.01; ***p<0.001. (B) MLE-12 cells were pretreated with a PKC inhibitor, bisindolylmaleimide (10 µM) for 1 hour prior to challenge with heat-inactivated PA as in (A) and cells were incubated for 2 hours. Cell lysates (30 µg proteins) were analysed by western blot analysis for histone acetylation. Shown is a representative blot of three independent experiments in triplicate. (C) Primary human bronchial epithelial cells (~60% confluence) were transfected with scrambled or PKC δ siRNA (50 nM) for 48 hours prior to PA challenge as in (A) for 1 and 3 hours. Cell lysates (20 µg proteins) were western blotted with acetylated histones H3 and H4, PKC δ, p-SPHK2, SPHK2 and total H3 and H4 antibodies. Shown is a representative western blot analysis from three independent experiments in triplicate. (D and E) MLE-12 cells grown to ~60% confluence on slide chambers were infected with DN PKC δ (25 MOI) for 24 hours prior to PA challenge for 1–3 hours. Cell were fixed, stained with p-SPHK2 antibody and phosphorylation of SPHK2 and its nuclear localisation was quantified by confocal microscopy. Original magnification, 60c, scale bar=20 µm. Shown is a representative confocal image (five areas per slide and four independent slides for each experiment). (E) Quantification of nuclear p-SPHK2 localisation in (D) by ImageJ. ***P<0.001. CFU, colony-forming unit; DAPI, 4’,6-diamidino-2-phenylindole; DN, dominant negative; MLE-12, murine lung epithelial-12; MOI, multiplicity of infection; PA, Pseudomonas aeruginosa; PKC, protein kinase C; SPHK2, sphingosine kinase 2.

PKC δ activation regulates PA-mediated IL-6 secretion and nuclear S1P levels. (A) MLE-12 cells (~60% confluence) were infected with control adenoviral or dominant negative (DN) adenoviral PKC δ (25 MOI) for 24 hours prior to challenge with heat-inactivated PA (1×108 CFU/mL) for varying time-periods, and IL-6 was measured in the supernatant. Values are means±SE form two independent experiments in triplicate. *P<0.05, **p<0.01; ***p<0.001. (B) MLE-12 cells grown on 100 mm dishes (~60% confluence) were transfected with scrambled or PKC δ siRNA (50 nM) for 48 hours prior to PA challenge as in (A) for 1 and 2 hours. ChIP assays were performed as described in ’Materials and methods' section. Data are means±SE from three independent experiments in triplicate. ***P<0.001. (C) Primary HBEpCs in 100 mm dishes (~60% confluence) were infected with control or DN PKC δ adenoviral vector (25 MOI) for 48 hours or cells were pretreated with ABC294640, a SPHK2 inhibitor for 1 hour prior to PA challenge. Nuclei were isolated, and lipids were extracted as outlined in ’Materials and methods' section. S1P levels in the nuclear fraction was determined by LC-MS/MS and quantified. Values are means±SE of two independent experiments in triplicate. *P<0.05, **p<0.01; ***p<0.001. ChIP, chromatin immunoprecipitation; CFU, colony-forming unit; DN, dominant negative; HBEpCs, human bronchial epithelial cells; IL-6, interleukin 6; LC-MS/MS, liquid chromatography tandem-mass spectrometry; MLE-12, murine lung epithelial-12; MOI, multiplicity of infection; PA, Pseudomonas aeruginosa; PKC, protein kinase C; S1P, sphingosine-1-phosphate; SPHK2, sphingosine kinase 2.
PA enhanced association of SPHK2 and PKC δ with HDAC1/2 in epithelial cell nucleus
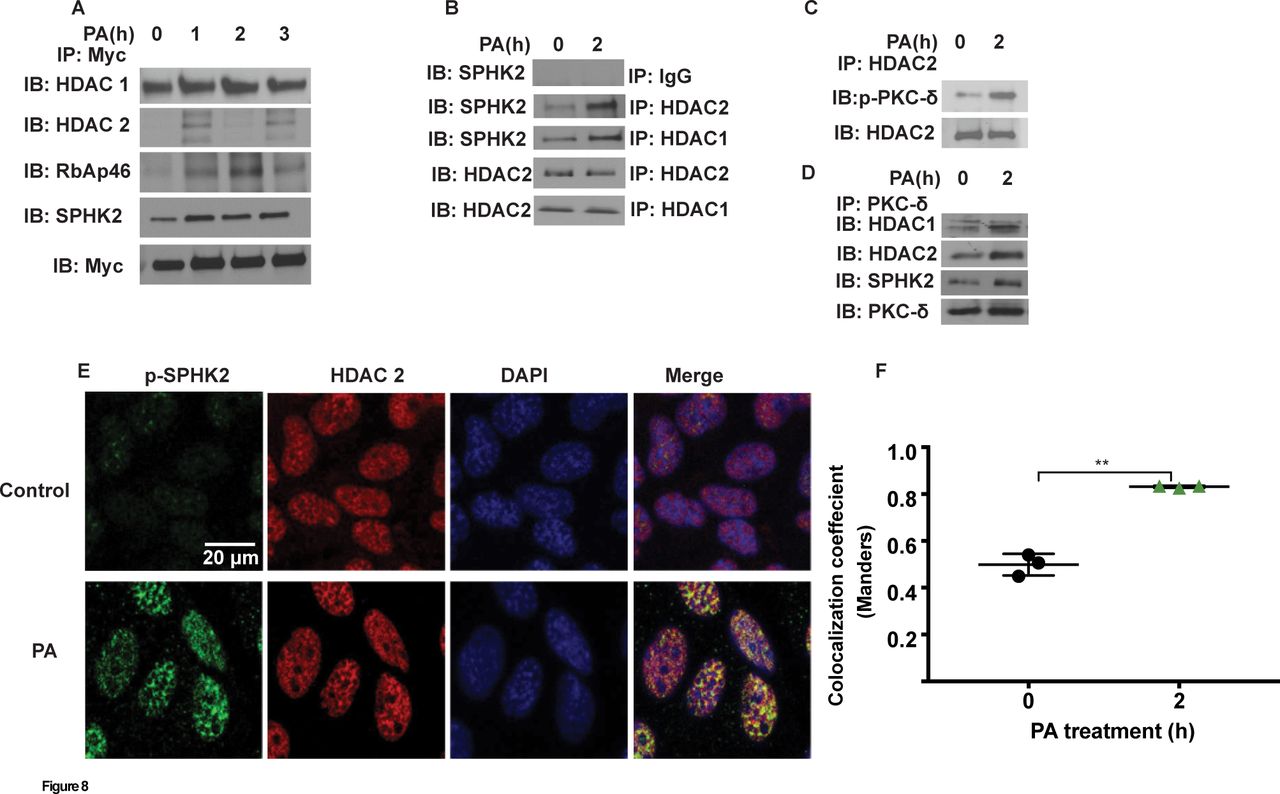
Having established PKC δ-dependent nuclear localisation of p-SPHK2 by PA in lung epithelial cells, next we investigated potential association between SPHK2 and PKC δ with HDAC1/2 in the nucleus. MLE-12 cells were infected with myc-tagged adenoviral Sphk2 WT construct for 24 hours prior to treatment with heat inactivated PA for 1–3 hours, cell lysates were immunoprecipitated with anti-myc antibody and immunoblotted. As shown in figure 8A, PA enhanced association of SPHK2 with HDAC1/2, and also with RbAp46/48, a component of Sin3 and NuRD co-repressor complexes. Similarly, immunoprecipitation of nuclear lysates from control and PA-treated MLE-12 cells with anti-HDAC1 or anti-HDAC2 antibody showed enhanced association between HDAC1/HDAC2 and SPHK2 (figure 8B) or p-PKC δ (figure 8C). Furthermore, immunoprecipitation of nuclear lysates with anti-HDAC2 or anti-PKC δ antibody revealed increased association of HDAC2 with PKC δ (figure 8C), and PKC δ with SPHK2, HDAC1 and HDAC2 (figure 8D). Immunohistochemical localisation also revealed enhanced association between p-SPHK2 with HDAC1 after PA challenge of MLE-12 cells (figure 8E & F). The above results suggested an enhanced association between SPHK2, HDAC1/2 and PKC δ in the nucleus after PA infection of epithelial cells. However, it is unclear if these interactions are direct or indirect involving other scaffolding proteins.

PA enhances association of SPHK2 with HDAC1/2 and S1P generation in the nucleus. (A) MLE-12 (~70% confluence) were infected with control adenoviral or myc-tagged SPHK2 WT adenoviral construct (25 MOI) for 25 hours prior to exposure to heat-inactivated PA (1×108 CFU/mL) for 1–3 hours. Cells were trypsinised, nuclei were isolated as described in ’Materials and methods' section, and nuclear extracts (1 mg protein) were subjected to immunoprecipitation with anti-myc antibody and immunoprecipitates were analysed for HDAC1, HDAC2, RBAP46, total SPHK2 and myc by western blot analysis. Shown is a representative blot from three independent experiments. (B), MLE-12 (~90% confluence) were challenged with PA as in (A) for 2 hours and at the end of the incubation period, cells wells trypsinised and nuclei were isolated as described in ’Materials and methods' section. Nuclear extracts (1 mg protein) were subjected to immunoprecipitation with IgG, anti-HDAC1 or anti-HDAC2 antibodies and immunoprecipitates were analysed for HDAC1, HDAC2 and total SPHK2 by western blot analysis. (C) Same as (B) and nuclear extracts (1 mg protein) were subjected to immunoprecipitation with anti-HDAC2 antibody and immunoprecipitate was analysed for HDAC2, and p-PKC δ by western blot analysis. (D) Nuclear extracts (1 mg protein) were subjected to immunoprecipitation with anti-PKC δ antibody and immunoprecipitates were analysed for HDAC1, HDAC 2, SPHK2, and PKC δ by western blot analysis. (E and F) MLE-12 cells grown on slide chambers (~90% confluence) and were challenged PA as in (A) for 2 hours. Cells were fixed and co-immunostained with anti-p-SPHK2 and anti-HDAC2 antibodies, fluorescence image stacks (30–40 focal planes with 0.5–1 µm spacing) was acquired by confocal microscopy. Original magnification, 60x, scale bar=20 µm. Shown is a representative micrograph from three independent experiments and co-localisation (F) was quantified by coloc-2 plugin in ImageJ (five areas per slide; four slides per group). **P<0.01. CFU, colony-forming unit; DAPI, 4’,6-diamidino-2-phenylindole; HDAC, histone deacetylase; MLE-12, murine lung epithelial-12; MOI, multiplicity of infection; PA, Pseudomonas aeruginosa; PKC, protein kinase C; RBAP, retinoblastoma-binding protein; S1P, sphingosine-1-phosphate; SPHK2, sphingosine kinase 2.
SPHK2 is localised in the nucleus of cystic fibrosis lung specimens
Patients with CF are plagued with chronic PA lung infection, a hallmark of the disease. The role of S1P and S1P metabolising enzymes in CF pathophysiology is unclear. Immunostaining of normal and CF lung tissue specimens with antiphospho SPHK2 antibody revealed extensive nuclear staining of p-SPHK2 in airway and alveolar epithelial cells in CF specimens compared with normal (figure 9A-K). On the other hand, normal lungs showed less localisation of p-SPHK2 in the nucleus of airway and vascular wall cells, and expression in alveolar septal cells was also less prominent than that found in CF samples and more localised staining in endothelial and smooth muscle cells of pulmonary artery.
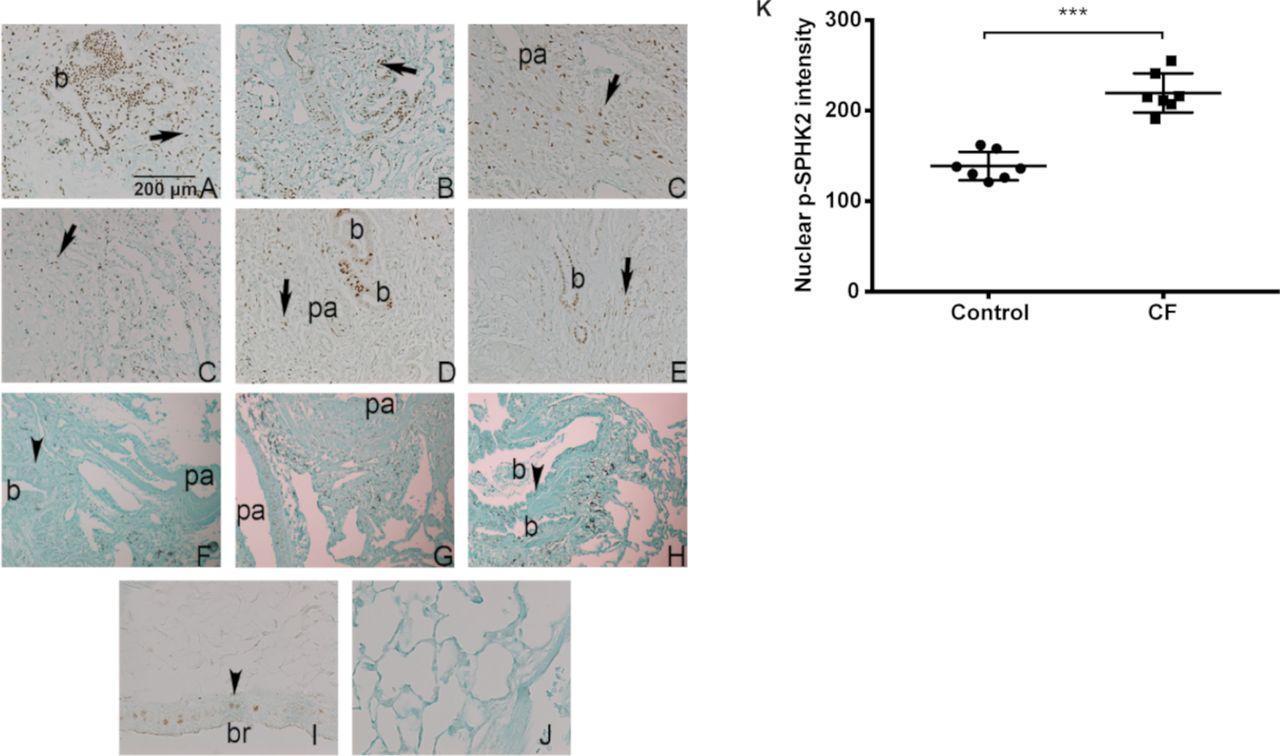
Nuclear localisation of p-SPHK2 in CF lung specimens. (A–E) Representative fields of six explanted CF lungs showing extensive nuclear SPHK2 staining in airway cells (b) and alveolar cells (arrows). There was more localised staining in endothelial and smooth muscle cells of pa. Scale bar=200 µm. (F–G) On the other hand, normal lungs (n=6) showed localised SPHK2 nuclear expression in airway (b) (arrowheads) and vascular wall cells. Expression in alveolar septal cells was also less prominent that that found in CF samples (G). Positive (I) and knockout negative (J) control mouse lungs (magnification bar=1 00 µm). (K) quantification of nuclear p-SPHK2 localisation by ImageJ. ***P<0.001. CF, cystic fibrosis; pa, pulmonary artery; SPHK2, sphingosine kinase 2.
Discussion
Sphingolipids and its metabolites have been implicated in normal cell functions and in the pathogenesis of human diseases including pulmonary disorders.16 39 40 Among the sphingolipid metabolites, ceramide plays an important role in bacterial infections,41 COPD42 and CF.43–45 Ceramide is central in sphingolipid rheostat as it can generate sphingosine, which is subsequently converted to S1P by SPHKs 1 and 2 by lung and other cells. We previously demonstrated that SPHK1/S1P signalling axis contributes to the development of several lung pathologies.19 21–23 29 35 36 While pathological increase of ceramide levels in the lung after PA infection was shown to be associated with apoptosis and cytokine responses, S1P was effective in clearing the mycobacterial burden in a mouse model of Mycobacterium tuberculosis infection.46 PA infection enhanced nuclear localisation of p-SPHK2 in alveolar and bronchial epithelial cells of the mouse lung (figure 4), and our current observation of predominant nuclear localisation of p-SPHK2 in alveolar and bronchial epithelial cells in CF lung specimens suggest a potential role of nuclear SphK2 in lung inflammation associated with CF. The role of SPHK2 in bacterial lung inflammatory injury was not restricted to PA, a Gram-negative bacterium as Sphk2-/- mice also conferred protection against pulmonary infection with methicillin-resistant Staphylococcus aureus (MRSA), a Gram-positive bacterium, compared with wild type and Sphk1-/- mice (online supplementary figure 7A and B). Our studies show for the first time a role for SPHK2-mediated nuclear S1P in PA-mediated lung inflammatory injury via epigenetic modification of pro-inflammatory genes, while blocking SPHK1 had no effect on PA-induced lung inflammation in vivo and H3 and H4 histone acetylation in lung epithelium. Thus, S1P generated by SPHK2 in the nucleus has a different role compared with S1P generated by SPHK1 in the cytoplasm in mediating the inflammatory response. Furthermore, our working model is that S1P generated in the cytoplasm/plasma membrane by SPHK1 is transported to the outside of the cell via a transporter such as Spns2 and signals via S1P1-5 in an autocrine or paracrine fashion. On the contrary, S1P generated in the nucleus by SPHK2 does not signal via S1PRs but modulates HDAC1/2 activity and brings about epigenetic changes and inflammatory responses. In addition to PA, MRSA also stimulated phosphorylation of SPHK2 and histone acetylation in vitro and genetic deletion of Sphk2 in mice reduced MRSA-induced lung inflammation (online supplementary figure 7) suggesting a common mechanism of sphingolipids and sphingolipid signalling in bacterial lung inflammatory injury.
A salient feature of this study is the ability of PA to phosphorylate host SPHK2 and generate S1P in a spatiotemporal manner in the nucleus of lung epithelial cells. Under basal condition, S1P levels are very low in nuclear preparations from lung epithelial (~10 pmol/mg protein) and breast cancer cell line Michigan Cancer Foundation -7(MCF-7) (~2 pmol)47compared with other sphingolipid bases. Infection of mouse lung or lung epithelial cells with PA-stimulated SPHK2 phosphorylation at ser/thr residue and its nuclear localisation, and inhibition of SphK2 activity with ABC294640, a specific inhibitor of SPHK2, reduced S1P accumulation in the nucleus. Both SPHK1 and SPHK2 are phosphorylated at ser/thr residues by ERK1/236 and PKC.47 PA infection of lung epithelial cells stimulated PKC activity and blocking PKC δ activity with DN Pkc δ or knockdown of Pkc δ with a siRNA attenuated SPHK2 phosphorylation in the nucleus, S1P production and IL-6 secretion suggesting an important role for this PKC isoform in nuclear S1P accumulation after bacterial infection of epithelial cells. Exposure of mouse embryonic fibroblasts to FTY720, an analogue of sphingosine, elevated FTY720-P levels both in the cytoplasm and nuclear fractions48; however, it is unclear if FTY720-P was generated in the nucleus. Recent evidences suggest that elevated nuclear S1P and S1P analogues are linked to increased risk for neural tube defects49 and neurodegeneration in neuron models of Huntington’s disease.50
Acetylation of lysine residues in histones is an epigenetic mark of active gene expression. The interplay between HATs and HDACs along with other chromatin remodelling factors tightly regulates gene expression. HDAC1 and HDAC2 are the core catalytic components of multiprotein repressor complexes, Sin3 and NuRD that regulate the chromatin structure in response to external stimuli. Previously, phorbol myristate acetate (PMA) induced SPHK2 localisation with nuclear HDACs1 and 2 in breast cancer MCF-7 cell line was shown to inhibit HDAC activity through nuclear S1P.47 It is intriguing to note in our studies that activated SPHK2 regulated the acetylation of lysine 9 in histone H3 and lysine 8 in histone H4, markers of active transcription, in mouse lung and epithelial cells, which was blocked by SPHK2 inhibitor. We also noted that the activation of PKC δ was paramount in downstream histone acetylation and inflammatory cytokine generation. While it has been reported earlier on the potential interaction between HDACs and SPHK2 and nuclear S1P inhibiting the HDAC activity in PMA-induced cancer cells,47 our study for the first time has shown such an interaction occurs during a bacterial infection in the lung epithelium. We observed that SPHK2/p-SPHK2 to be an integral component of the nuclear HDAC repressor complex Sin3 and NuRD and blocking SPHK2 or PKC δ inhibited PA-induced H3 and H4 histone acetylation. We also observed a significant increase in the interaction between HDAC1/2 and p-SPHK2 after PA treatment and this interaction was crucial in regulating the spatiotemporal generation of S1P in the nucleus. While the mechanism by which S1P generated in the nucleus regulates HDAC activity is still an open question, molecular modelling of S1P binding into the active site of HDAC1 homologue from Aquifex aeolicus showed that S1P was docked tightly in the pocket with the highly conserved Arg27 with a predicted binding energy that was comparable to the binding energy of HDAC inhibitor, suberoylanilide hydroxamic acid.47 These results suggested that binding of S1P, similar to HDAC inhibitors, could directly inhibit HDAC activity.
Persistent PA infection in patients with CF lead to rapid deterioration of lung function leading to increased morbidity rates.51 Loss of CFTR promotes a favourable environment to PA to exert its pathogenic effect by rapid mutations leading to antibiotic resistance.52 While there is no consensus on whether the elevated pro-inflammatory cytokines, like IL-1, IL-6, IL-8 and TNF-α, in patients with CF is secondary to PA infection or due to the loss of CFTR, they promote influx of neutrophils into the airway leading to progressive airway damage. Chronic PA infection in CF cannot be eradicated by antibiotics and ameliorating lung inflammation by targeting SPHK2 and nuclear S1P signalling may supplement the host innate immunity, offering a new paradigm in the management of inflammation of the CF lung.
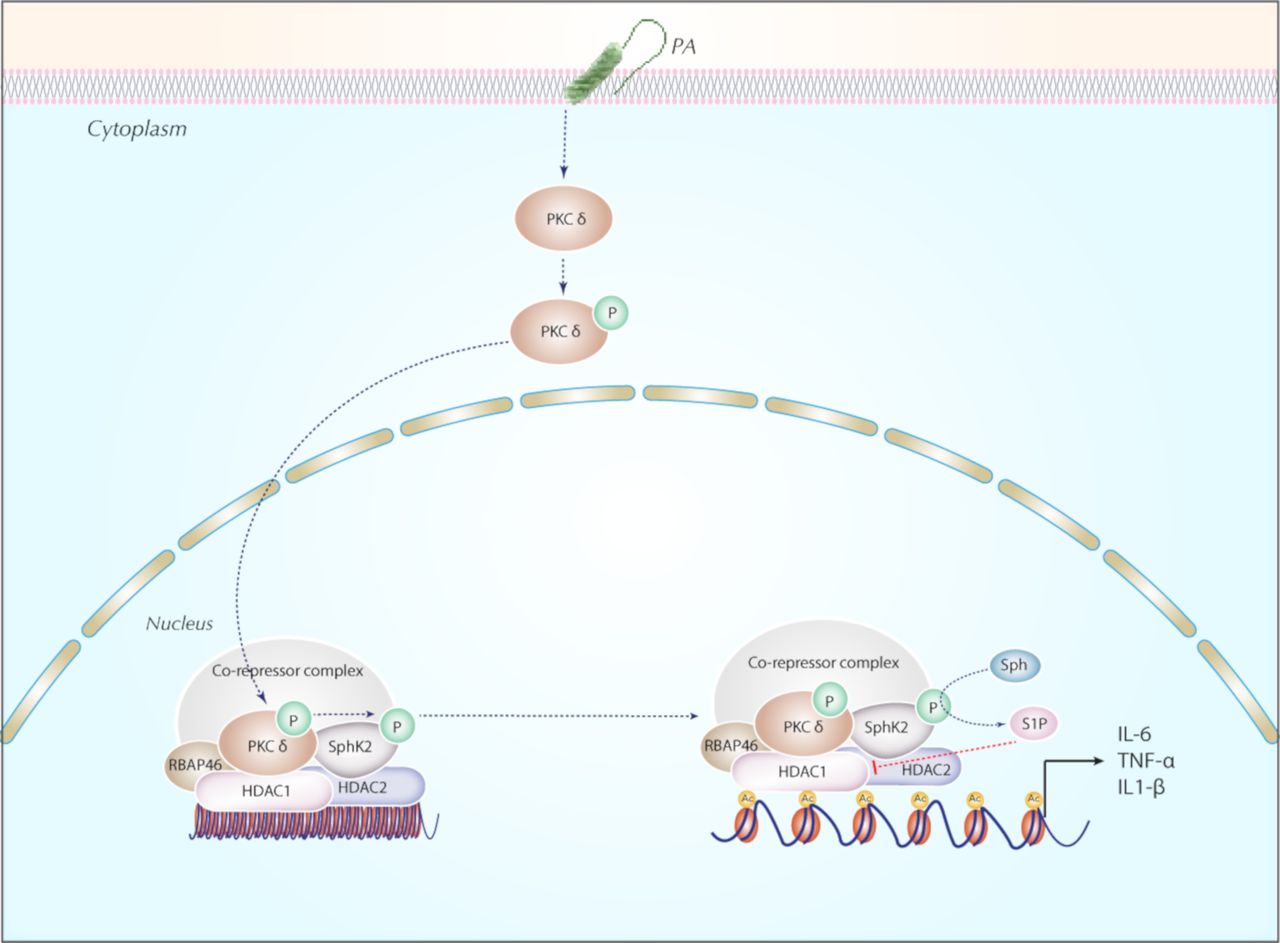
In summary, our findings collectively support a novel role for nuclear SPHK2/S1P signalling in secretion of pro-inflammatory cytokines secretion via modulation of HDAC1/2 activity (figure 10). The anti-inflammatory effect of inhibition of PKC δ and SPHK2 described here suggested that targeting PKC δ and/or SPHK2 might be a potentially useful therapeutic strategy in ameliorating bacterial lung inflammatory injury.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model for the role of nuclear SPHK2/S1P signalling axis in the epigenetic regulation of PA-induced lung inflammatory injury. In lung epithelium, PA infection phosphorylates SPHK2 and its nuclear localisation through PKC δ activation generating nuclear S1P. Nuclear S1P modulates HDAC1/2 activity that leads to chromatin plasticity and increased expression of pro-inflammatory cytokines, IL-6 and TNF-α. Blocking PKC δ, and/or SphK2 attenuated PA-mediated lung inflammatory injury. HDAC, histone deacetylase; IL-6, interleukin 6; PA, Pseudomonas aeruginosa; PKC, protein kinase C; RBAP, retinoblastoma-binding protein; S1P, sphingosine-1-phosphate; SPHK2, sphingosine kinase 2; TNF-α, tumour necrosis factor α.
Acknowledgments
The authors would like to thank Dr Prasad Kanteti for helpful discussions and Dr Richard Proia from National Institutes of Health for Sphk1 and Sphk2 deficient mice. The authors would also like to thank the VCU Lipidomics/Metabolomics Core, the NIH-NCI Cancer Center Support Grant P30 CA016059 to the VCU Massey Cancer Center as well as a shared resource grant (S10RR031535) from the National Institutes of Health.
References
Footnotes
VN and PF contributed equally.
Contributors PF, DLE, VN, CT, YK, and EVB designed the research; DLE, EVB, IAB, VS, AWH, AH, RMT and VN performed the experiments; VN, DLE and PF analyzed the data and wrote the manuscript.
Funding This work was partly supported by the US National Institutes of Health grant P01 Hl09850 to VN.
Competing interests None declared.
Ethics approval This study was approved by the University of Colorado Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it was published Online First. A correction was made to Figure 4.