Article Text

Abstract
Introduction Prognostic biomarkers have been very elusive in the lung squamous cell carcinoma (SCC) and none is currently being used in the clinical setting. We aimed to identify and validate the clinical utility of a protein-based prognostic signature to stratify patients with early lung SCC according to their risk of recurrence or death.
Methods Patients were staged following the new International Association for the Study of Lung Cancer (IASLC) staging criteria (eighth edition, 2018). Three independent retrospective cohorts of 117, 96 and 105 patients with lung SCC were analysed to develop and validate a prognostic signature based on immunohistochemistry for five proteins.
Results We identified a five protein-based signature whose prognostic index (PI) was an independent and significant predictor of disease-free survival (DFS) (p<0.001; HR=4.06, 95% CI 2.18 to 7.56) and overall survival (OS) (p=0.004; HR=2.38, 95% CI 1.32 to 4.31). The prognostic capability of PI was confirmed in an external multi-institutional cohort for DFS (p=0.042; HR=2.01, 95% CI 1.03 to 3.94) and for OS (p=0.031; HR=2.29, 95% CI 1.08 to 4.86). Moreover, PI added complementary information to the newly established IASLC TNM 8th edition staging system. A combined prognostic model including both molecular and anatomical (TNM) criteria improved the risk stratification in both cohorts (p<0.05).
Conclusion We have identified and validated a clinically feasible protein-based prognostic model that complements the updated TNM system allowing more accurate risk stratification. This signature may be used as an advantageous tool to improve the clinical management of the patients, allowing the reduction of lung SCC mortality through a more accurate knowledge of the patient’s potential outcome.
- signature
- immunohistochemistry
- squamous cell carcinoma
- disease-free survival
- overall survival
- clinical utility
- prognosis
Statistics from Altmetric.com
- signature
- immunohistochemistry
- squamous cell carcinoma
- disease-free survival
- overall survival
- clinical utility
- prognosis
Key messages
What is the key question?
In the clinical settings, no prognostic biomarkers are used for risk stratification in patients with early stage lung squamous cell carcinoma (SCC).
What is the bottom line?
We describe and validate a clinically feasible 5-protein-based prognostic signature that complements the updated TNM system allowing more accurate risk stratification of patients with SCC.
Why read on?
We used robust methodological and statistical methods to describe the signature, and validated the results in three independent retrospective cohorts of patients with SCC.
Introduction
Lung cancer remains the leading cause of cancer-related death worldwide.1 Non-small cell lung cancer (NSCLC) is subdivided into two main histological subtypes, adenocarcinoma (ADC) and squamous cell carcinoma (SCC), accounting for 50% and 30% of NSCLC cases, respectively.2 The tumour-node-metastasis (TNM) staging system is currently the most valuable prognostic indicator to guide treatment decisions for both ADC and SCC.3 However, this classification procedure is not totally accurate since different disease progression and survival outcomes in patients within each individual stage are commonly observed. In this regard, there is an urgent medical need to identify new markers that help to identify those patients at high risk of recurrence in each surgically amenable (I–III) stage. A precise prognostic indication is especially relevant to help the clinician’s decision to recommend or not adjuvant therapy, currently based on classical chemotherapies, but also in the future on molecular targeted drugs or immune checkpoint inhibitors. Moreover, the need for improving the prognostication tools and models is increasing, since the implementation of lung cancer screening programmes will undoubtedly elevate the proportion of patients with early-stage NSCLC undergoing surgery.
During the last decades, high-throughput genomic tools have enabled the identification of RNA-based diagnostic and prognostic models with potential clinical value.4–7 Despite the efforts, the clinical utility and reproducibility of most of these signatures are under debate.8 Importantly, although two real-time PCR-based signatures are in a more advanced developmental stage for ADC,4 5 no molecular-based signature is clinically available for SCC. We have recently identified and validated the clinical utility of a three protein-based prognostic signature to stratify patients with early lung ADC according to their risk of recurrence or death. Also, we have demonstrated that this signature adds further prognostic information to the eighth version of the TNM-based clinical staging. More importantly, it may be a valuable tool to select stage I–IIA patients who could obtain a benefit from adjuvant chemotherapy.9 In the case of the SCC subtype, some prognostic signatures have been published, but most of them lack an independent validation cohort.10–12 Therefore, none has been incorporated into the clinical practice. Modelling of prognostic factors in SCC is a more difficult task as compared with ADC likely due to a broader intrinsic biological heterogeneity.13
In the present work, we have developed and validated a simple, feasible and reproducible protein-based prognostic signature for surgically amenable lung SCC. The use of proteins as markers in our signature poses a series of advantages for clinical application in comparison with high throughput technologies. Protein determination is still more accessible and straightforward in the routine clinical setting in health-providing centres. Our signature readily identifies early-stage patients with high and low risk of recurrence or death independently of TNM and provides complementary information to this gold standard staging system. In fact, the prognostic discriminating power is significantly improved when the protein-based prognostic signature is combined with the TNM staging system. Therefore, the molecular classifier proposed in this study may be very useful to refine the current prognostic information and could be used together with TNM to tailor more accurately the management of patients with SCC after surgery.
Material and methods
Patients
Primary SCC samples for this study were obtained from surgical specimens from University of Texas MD Anderson Cancer Centre (Houston, Texas, USA) (MDA), the multicentre randomised NATCH trial,14 the multi-institutional Pulmonary Biobank Platform (Spain) (CIBERES) and Clinica Universidad de Navarra (Pamplona, Spain) (CUN). SCC tumours were classified according to the WHO 2004 classification15 and the eighth TNM edition was used for tumour stratification,16 except for the NATCH cohort in which, due to the time when the cohort was collected, the patients were staged according to the sixth edition of the IASLC staging method. Inclusion criteria were as follows: patients with SCC histology, complete resection of the primary tumour and absence of chemotherapy or radiotherapy treatment prior to surgery. Patients diagnosed with mixed or combined SCC histology, previous lung cancer or synchronous lung tumours were excluded. The MDA cohort was composed of 117 patients with SCC diagnosed from 1999 to 2008 at the MD Anderson Cancer Centre (Houston, Texas, USA). Two different cohorts were used for the validation, the NATCH cohort (n=96) and the CIBERES-CUN cohort (n=105, 63 patients from CIBERES multi-institutional Pulmonary Biobank Network and 42 patients from CUN). Disease-free survival (DFS) and overall survival (OS) were calculated from the date of surgery to the date of recurrence or death, respectively. For survival analyses, the follow-up period was restricted to 60 months. Reported recommendations for tumour marker prognostic studies (REMARK) criteria were followed throughout the study.17 The study was conducted according to the Declaration of Helsinki. Written informed consent was obtained from each patient. Characteristics of the cohorts are specified in the online supplementary table 1.
Supplemental material
Experimental procedures
In silico analysis, cell culture, western blotting, and immunohistochemical staining and quantification were performed as previously described.9 Briefly, immunohistochemical staining was performed on TMAs from formalin-fixed paraffin-embedded tissues and cell block sections for the detection of the following proteins: BRCA1, CDC6, LIG1, QKI, RAD51, RAE1, RRM2, SIRT2, SLC2A1, SNRPE, SRSF1 and STC1. Antibody characteristics are summarised in the online supplementary table 2. The specificity of each antibody was thoroughly assessed using Western blotting, immunohistochemistry (IHC) and siRNA knock-down technology in NSCLC cells (online supplementary figure 1).
Statistics
TRIPOD criteria were followed in our study18 and statistical analyses were performed using SPSS 22.0 and STATA/IC 12.1. Development of the prognostic signature was carried out in the MDA cohort by regression Cox analysis by steps for DFS where the contribution of every potential predictor to the model performance was assessed manually by the researcher. We introduced all protein expression-based variables into the Cox analysis and eliminated redundant variables one by one according not only to their significance order (from lower to higher significance) but also avoiding the loss of >10% of the initial magnitude of the model χ2. We formulated a prognostic index (PI) for the model as the sum of the products of the B coefficients for each variable and its expression value (H-score). Discriminative ability of the PI was assessed by Harrell’s Concordance coefficient (C-index). The models were also evaluated by Kaplan-Meier curves and log-rank test for both DFS and OS, and stratifying the PI by the upper tertile into two groups of risk (low and high). As a complementary study, the signature was also assessed stratifying the patients by the median. The model (C-index calculation) was internally validated by bootstrapping following Harrell’s guidelines.19 Specifically, the bootstrapping method was used to quantify any optimism in the predictive performance through a shrinkage penalisation strategy. Univariate and multivariate Cox proportional hazards analyses including other clinical and pathological variables were used to assess the prognostic role of the molecular model (PI). Variables with p<0.25 in the univariate analysis were included in the multivariate analysis. Additivity was assessed by verifying that all pairwise interaction terms were clearly non-significant, and, for quantitative predictors, linearity by testing the non-significance of squared terms. The model was externally validated in two cohorts, NATCH and CIBERES-CUN cohorts. Clinical utility of the model was analysed by comparing the likelihood ratio of the stage alone with that after addition of the molecular model (PI) through a bivariable Cox analysis. Moreover, we developed a new combined prognostic index (CPI) model by Cox regression, adding the pathological stage and the molecular data following this formula: (PI * B coefficient PI)+B, where B is a coefficient that changes for each stage. Also, the discriminative ability of the CPI was assessed as described above (Harrell’s C and the log-rank test for the CPI dichotomized at the upper tertile or the median). The combined model was validated in the CIBERES-CUN cohort. Univariate and multivariate Cox proportional hazards analyses including other clinical and pathological variables were used to assess the prognostic role of the CPI.
Results
Selection of genes related to clinical outcome in NSCLC
With the aim of generating a protein-based classifier to predict survival in patients with lung cancer , we selected genes associated with clinical outcome in the literature, as previously described.9 A flowchart with all the steps followed in this work is shown in figure 1. Briefly, we chose genes significantly associated with prognosis in at least two previously published mRNA-based expression signatures (20 genes out of 967). Moreover, nine additional biomarkers were added to the list, based on our previous results. Next, an in silico analysis using data from nine different databases was performed to check the prognostic value of these 29 genes at mRNA in NSCLC (online supplementary figure 2). A total of 21 biomarkers were associated with prognosis in at least two databases. Later, the availability and reliability of commercial antibodies were analysed. Nine genes were discarded at this point (five lacked reliable commercial antibodies and four genes whose antibodies did not demonstrate specificity). We finally selected 12 cancer-related genes (BRCA1, CDC6, LIG1, QKI, RAD51, RAE1, RRM2, SIRT2, SLC2A1, SNRPE, SRSF1 and STC1) and developed the prognostic model.

Organisation chart showing the main steps followed in this study. We first performed a literature search of genes with prognostic relevance in different lung cancer prognostic signatures or genes with prognostic value itself. We studied the prognostic value of each gene individually at RNA level by using nine expression array databases. After selecting genes with significant prognostic value in at least two databases, we searched for cancer hallmarks previously associated with each gene, discarding genes unrelated with the carcinogenic process. We analysed the specificity of the antibodies and selected those that passed our requirements. To develop the signature, we first studied the expression of the 12 selected proteins in the training cohort (MDA cohort) and established a parsimonious prognostic model by Cox regression (PI model). The model was externally validated in two independent cohorts of patients with lung SCC (NATCH and CIBERES-CUN). We finally studied the clinical usefulness of the model comparing its prognostic ability with the gold-standard TNM and combining both parameters (CPI model). CPI, combined prognostic index; IHC, immunohistochemistry; PI, prognostic index; SCC, squamous cell carcinoma; TNM, tumour-node-metastasis; WB, Western blotting.
Development of a protein-based signature for risk stratification in SCC
We first evaluated the expression and subcellular localisation of each of the selected 12 proteins (N, nuclear; C, cytoplasmic; MB, membrane) in the training cohort (MDA). Representative images of primary tumours are shown in figure 2. We next performed a Cox regression analysis to develop a parsimonious protein prognostic model based on the expression of these proteins. We selected the best model according to a high C-index coefficient and high parsimony (ie, the most robust and simplest model). The model was composed of five different proteins (RAE1, RRM2, SLC2A1, SRSF1 and STC1) divided into six variables (RAE1C, RRM2C, SLC2A1C, SRSF1N, STC1C and STC1N). The algorithm was: PI=−0.012 × H-Score RAE1C + 0.009 × H-Score RRM2C + 0.006 × H-Score SLC2A1C + 0.013 × H-Score SRSF1N + 0.011 × H-Score STC1C - 0.009 × H-Score STC1N. C-index was 0.68 for DFS and 0.61 for OS. The PI fulfilled the proportional hazards assumption (p=0.177). Stratifying by the upper tertile, the predicted low-risk group had significantly longer DFS and OS when compared with the high-risk group (figure 3A; p<0.001 and p=0.002, respectively). Interestingly, we also observed significant differences in the outcome when we stratified patients by the median for both DFS and OS (online supplementary figure 3A; p=0.008 and p=0.166, respectively).
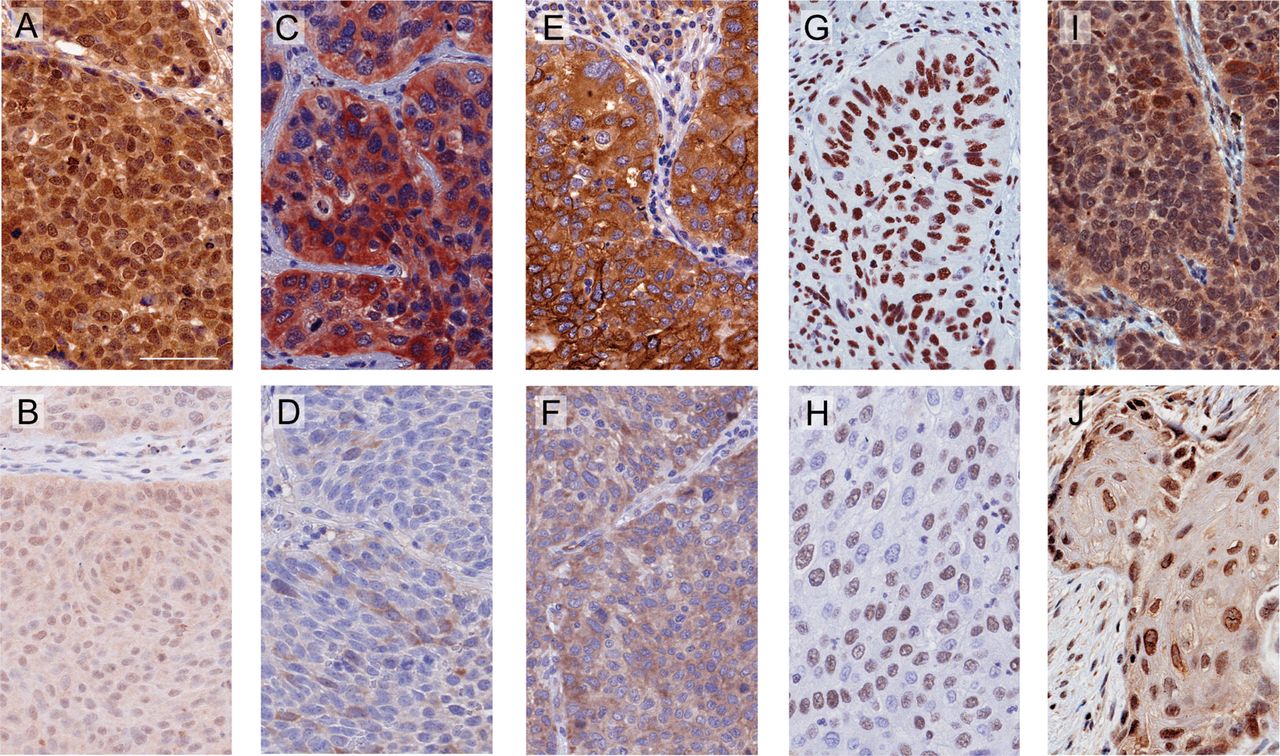
Representative immunostaining for the proteins included in the prognostic signature. Representative images were selected from tumour tissues with high (top) and low (down) expression of the proteins. (A and B) RAE1 appeared in both nuclear and cytoplasmic compartments. (C and D) RRM2 was found in the cytoplasm. (E and F) SLC2A1 was located in the cytoplasm and cell membrane. (G and H) SRSF1 was found specifically in the nucleus. (I and J) STC1 was detected in both nuclear and cytoplasmic compartments. Scale bar: 60 µm.

Association between PI and survival in patients with SCC. (A) Kaplan-Meier survival curves for high and low PI groups in MDA patients for DFS (left panel) and OS (right panel). (B) Kaplan-Meier survival curves for high and low PI groups in NATCH validation cohort for DFS (left panel) and OS (right panel). (C) Kaplan-Meier survival curves for DFS in CUN (left panel) and for OS (right panel) in CIBERES-CUN validation cohort. Patients were stratified into two risk groups according to the upper tertile of the PI. In all cases, differences between groups were evaluated using the log-rank test. DFS, disease-free survival; OS, overall survival; PI, prognostic index.
The PI was associated with prognosis of the 5-year outcome for both DFS (p<0.001) and OS (p=0.003) in a univariate Cox proportional hazards analysis (table 1). We next adjusted the PI with the stage in a multivariate Cox analysis. The molecular PI remained a significant predictor DFS (p<0.001; HR=4.06, 95% CI 2.18 to 7.56) and OS (p=0.004; HR=2.38, 95% CI 1.32 to 4.31). Same results were found when we analysed the PI without any categorisation (p<0.001 for DFS and p=0.008 for OS; online supplementary table 3).
Univariate and multivariate Cox proportional hazards analysis of PI and clinicopathological parameters for DFS and OS in the MDA cohort
Validation of the protein-based prognostic score
With the aim of analysing the potential transportability of our findings to other patient cohorts, we evaluated the prognostic potential of the model in two independent cohorts of patients with SCC by Kaplan-Meier analysis and log-rank test (NATCH and CIBERES-CUN, online supplementary table 1). The model was applied to the patients from NATCH cohort stratifying into two risk groups as above. Patients with high PI were significantly associated with poor survival (figure 3B; p=0.036 for DFS and p=0.020 for OS). In the CIBERES-CUN cohort, although DFS data were only available for 42 patients, high PI was also associated with shorter DFS (p=0.011, figure 3C) and tended to be associated with poor OS (p=0.050; figure 3C). In this cohort, the results were also significant when patients were stratified by the median (online supplementary figure 3B; p=0.031 and p=0.029, respectively).
We next performed a univariate and multivariate Cox regression analysis in the validation cohorts. In the multivariate analysis in the NATCH cohort, PI remained significant after adjusting for the clinicopathological variables (p=0.042 for DFS and p=0.031 for OS, online supplementary table 4). In the multivariate analysis of CIBERES-CUN cohort, a PI remained very close to the significance for DFS (p=0.050; HR=3.55, 95% CI 0.98 to 12.87) and OS (p=0.074; HR=1.84, 95% CI 0.94 to 3.60) when we stratified patients in two groups according to the upper tertile (online supplementary table 5).
As an indirect validation step, we performed an ‘inverse’ validation of the performance of our PI model by applying the protein algorithm to the mRNA expression levels of a fourth independent cohort, using the survival and mRNA expression data for SCC tumour samples from the TCGA database (n=334). Kaplan-Meier curves and log-rank test showed statistical differences when we stratified the patients into two groups according to the first tertile. Specifically, patients with SCC with high ‘mRNA PI’ exhibited shorter OS than low mRNA patients (p=0.043; online supplementary figure 4).
Clinical utility of the prognostic signature
To further assess the clinical relevance of the prognostic model, we examined whether the combination of the molecular PI and the pathological stage improved the predictability of the model in the MDA cohort. A bivariable Cox model was employed. As expected, stage was a strong prognostic factor for both DFS and OS (Likelihood ratio/χ2 test statistic (LR-χ²)=18.879, p<0.001 and LR-χ²=7.950, p=0.019, respectively). Nonetheless, there was a significant increase in the LR-χ² after adding the PI information (LR-χ²improvement=18.710, p<0.001 for DFS and LR-χ² improvement=6.436, p=0.011 for OS; online supplementary table 6). This significant increase reflected that the molecular information improves the prognostic information given just by the TNM-based stage.
Then we conducted a Cox regression analysis to generate a new prognostic model combining stage and the molecular model using the MDA cohort. This new signature was called CPI, and was formulated as follows: CPI=1.019 × PI+B; where B is a coefficient that changes for each stage (I, B=0; II, B=0.488; and III, B=1.523). The model performance was improved with the introduction of the stage information (C-indexCPI=0.74 for DFS; C-indexCPI=0.65 for OS). After CPI upper tertile stratification, the high-risk group showed significantly shorter DFS and OS (p<0.001 for both in the log-rank test; figure 4A). Moreover, a log-rank analysis stratifying the CPI according to the median yielded very similar results, supporting the robustness of the signature (online supplementary figure 3C). Besides, in a Cox regression analysis, the prognostic capacity of the CPI was independent of clinicopathological parameters using the CPI variable dichotomised by the upper tertile (p<0.001; HR=4.12, 95% CI 2.26 to 7.51 for DFS and p<0.001; HR=3.1, 95% CI 1.71 to 5.62 for OS; table 2) or using the CPI without stratification (p<0.001 for both DFS and OS; online supplementary table 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Assessment of the clinical utility of the prognostic signature in patients with SCC. (A) Association between CPI and survival in patients from MDA cohort. Kaplan-Meier survival curves were generated for DFS (left panel) and OS (right panel) (B). Validation of the CPI model. Kaplan-Meier curves for DFS (left panel) and OS (right panel) in CIBERES-CUN patients. Patients were stratified into two risk groups according to the upper tertile of the CPI. In all cases, differences between groups were evaluated using the log-rank test. CPI, combined prognostic index; DFS, disease-free survival; OS, overall survival; SCC, squamous cell carcinoma.
Univariate and multivariate Cox proportional hazards analysis of CPI and clinicopathological parameters for DFS and OS in MDA cohort
We further validated the prognostic value of the CPI model in the independent CIBERES-CUN cohort (stage I–III patients, n=104), for which eighth TNM data were available. In the case of DFS (CUN patients, n=41), PI addition significantly improved the risk stratification given by clinicopathological parameters (LR-χ²improvement=5.817, p=0.016; online supplementary table 6). We also observed the fact that those patients with high CPI were associated with shorter DFS, using the log-rank test (p=0.004, figure 4B). Moreover, after multivariate adjustment, CPI remained a prognostic factor for DFS (p=0.012; HR=5.52, 95% CI 1.46 to 20.89); online supplementary table 8). In the case of OS (n=104), the risk stratification was also improved when the PI was added to the stage (LR-χ² improvement=5.945, p=0.015, online supplementary table 6). Log-rank test showed a significant association between high CPI and shorter OS (p<0.001 using upper tertile as the cut-off, figure 4B; and p=0.001 using median, online supplementary figure 3D). After multivariate analysis, the CPI remained a significant predictor of 5 year OS (p=0.001; HR=2.88, 95% CI 1.51 to 5.49; online supplementary table 8). The analysis also yielded significant results when we used the CPI as a continuous variable, reinforcing the previous data (p=0.009 for DFS ad p<0.001 for OS; online supplementary table 9). Therefore, the data shown herein reveal that the molecular PI offers additional prognostic information to that provided by the mere clinicopathological stage.
Discussion
Lung SCC is the second most frequent histological subtype of NSCLC, and the overall 5-year survival rate of SCC shows still dismal figures. The identification of prognostic markers for SCC could complement the TNM criteria and assist in the clinical management of individual patients, allowing a more precise tool to discriminate which patients should be treated more thoroughly (ie, with adjuvant treatment) and which patients could avoid the treatment and thus, skip the potential toxicity from chemotherapeutic agents. Here, we identify a promising protein-based signature as a reliable tool for lung SCC prognostication.
There are two critical points that need to be carefully considered to ensure the validity of any prognostic model: transportability among different cohort settings and clinical utility.8 Our protein-based prognostic model overperforms other SCC prognostic signatures that have not been replicated in SCC-independent cohorts.6 10–12 20 21 Moreover, the fact that our validation cohort is already a multi-institutional cohort, including patients of different hospitals, suggests the applicability of the SCC signature to different types of patients and clinical settings.
Some prognosis-based SCC signatures have been validated22–24; however, the majority of them do not address the main practical issue of the medical utility.25–28 With the exception of Larsen et al,23 the rest of the studies lack a comparison between the performance of their models and the TNM.23 On the contrary, we have demonstrated not only that our model (PI) is able to identify a subset of patients with high risk of recurrence but also the ability of the signature to improve the prognostication value of the TNM through a CPI. Furthermore, we clearly show that our model yields additional information to the TNM and, thus, the integration of both molecular information and pathological stage refines the risk prediction of progression or survival in a more accurate manner. A similar approach was followed by Grinberg et al, combining the protein data with clinical parameters. However, their model failed to support the prognostic value in the validation cohort of patients with SCC.29
Another potential advantage of our model is that it is based on the expression of five proteins detected by IHC. Thus, our signature has several benefits when compared with other proposed signatures. First, it is based on proteins, the main biological molecules governing normal and pathological cell functions. Other protein-based signatures have been developed for NSCLC,29 30 which have failed in their validation in SCC histology; second, we analyse only five individual proteins to prognosticate the patients, while most of the previously proposed signatures are composed of dozens to hundreds of genes20–23; third, the detection of the proteins is performed by IHC, available in every hospital through their Diagnostic Pathology Services. In terms of routine clinical translation, the cost-efficiency and feasibility of the IHC technique provide advantages that may facilitate their implementation into the clinical practice, as has been already established for other companion biomarkers of novel personalised therapies.31 One of the advantages of IHC is that it allows the assessment of protein levels in specific subcellular compartments. It is well established that protein subcellular localisation has profound functional and prognostic effects for a large number of proteins.32 Together with the development of automatic programmes to quantify the staining, the experience in different fields supports the fact that new protein-based signatures may be easily implemented into the clinical practice in the near future.
The proposed model has been developed from a panel of genes previously related to lung cancer prognosis and carcinogenesis.4 5 21 22 33–44 Particularly, RAE1 and SRSF1 exert their function in RNA metabolism, a process which is dysregulated in lung cancer.45 Our study clearly supports previously published data showing that high SRSF1 associates with poor NSCLC prognosis.34 In our model, the cytoplasmic levels of RAE1 are associated with better survival in contrast to previous findings in ADC.33 These contrasting results may be explained by functional differences of the gene between the two histologies. Further analyses would be needed to clarify this issue. RRM2 is a ribonucleotide reductase which participates in DNA repair, invasion and proliferation and has been related to poor lung cancer prognosis.46 SLC2A1 is a glucose receptor (also named GLUT1) involved in the uptake of glucose by mammalian cells.47 Here, we confirm previous data showing that high levels of both proteins are associated with poor prognosis in lung cancer.48 49 Finally, stanniocalcin 1 (STC1) exerts important functions in tumourigenesis and metastasis.50 51 Here we show that the levels of this protein in the nucleus or the cytoplasm are related to opposite outcomes, suggesting different functions within the cell.
A limitation of this study is that, due to the number of cases, we have not been able to perform a specific statistical analysis for stage I patients, which have a special interest in terms of clinical management. A further additional validation study with a cohort including large numbers of patients with stage I SCC patients will help to solve this question in the future. Also, the inclusion criteria (SCC histology, complete resection and non-neoadjuvant patients) used in the study might affect the generalisability of the developed signature for future studies. Thus, further studies need to be performed to apply the model to different populations.
In summary, this study identifies and validates a five protein-based signature for patients with early lung SCC. The prognostic signature has been developed with robust methodological and statistical tools to guarantee their robustness and transportability. The PI stratifies the risk of recurrence or death accurately in different cohorts of patients with SCC and complements the TNM staging system. Most importantly, our data suggest that the CPI model could help physicians to accurately tailor the management of patients with SCC after surgery.
Acknowledgments
The authors thank Cristina Sainz and Amaya Lavín for technical assistance, Dr. Marta García-Granero for statistical support and all patients of the centers included in this study. We also thank the CIBERES multi-institutional Pulmonary Biobank Platform Network (Spain) for providing the material to validate the signature, specially: Ana Blanco (Hospital Virgen del Rocío, Sevilla), Leonardo de Esteban (Hospital Joan XXIII, Tarragona), Esther Fernández (Servicio de Cirugía Torácica, Hospital Universitari Germans Trias i Pujol, Badalona), Mª Jesús Fernández- (Fundación Jiménez Díaz, Madrid), Mª Ángeles González (Hospital Virgen del Rocío, Sevilla), Guadalupe González (Pont Hospital Mutua de Terrassa, Terrassa) , Pedro López de Castro (Hospital Joan XXIII, Tarragona and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), Carlos Martínez-Barenys (CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), José Luis Mate (Anatomía Patológica, Hospital Universitari Germans Trias i Pujol, Badalona), Rosario Melchor (Fundación Jiménez Díaz, Madrid), Ramón Rami-Porta (Hospital Mutua de Terrassa, Terrassa and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), Julio Sánchez de Cos (Servicio de Neumología, Hospital San Pedro de Alcántara, Cáceres and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), Rosa María Sánchez (Hospital Virgen del Rocío, Sevilla), Jaume Sauleda (Servicio de Neumología, Hospital Universitari Son Espases, Palma, Institut d’investigació Sanitària de Palma, IdISPa and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid,), Sergio Scrimini (Servicio de Neumología, Hospital Universitari Son Espases, Palma, Institut d’investigació Sanitària de Palma, IdISPa and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), Luis Seijo (Fundación Jiménez Díaz, Madrid and CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid), Mireia Serra (Hospital Mutua de Terrassa, Terrassa) y Cristina Villena (CIBER de Enfermedades Respiratorias – CIBERES, Instituto de Salud Carlos III, Madrid).
References
Footnotes
Contributors EMT, MJP and LMM: conception and design. All authors: acquisition of data; analysis and interpretation of data; writing, review, and/or revision of the manuscript.
Funding This work was supported by FIMA, Spanish Ministry of Economy and Innovation and Fondo de Investigación Sanitaria-Fondo Europeo de Desarrollo Regional (12/02040, PI13/00806, PI14/01678, PI 14/01686, PI16/01821, RTICC RD12/0036/0040, RD01/0036/0072), PII Oncologia Torácica SEPAR y Ciber de Enfermedades Respiratorias-CIBERES–FEDER, CIBERONC and AECC Scientific Foundation (GCB14-2170).
Competing interests JA, EMT, MJP, RP and LMM are listed as co-inventors of a recently applied patent related to some of the results included in this manuscript.
Patient consent Not required.
Ethics approval Institutional Review Board and Ethical committees of all participating institutions: Clínica Universidad de Navarra, MD Anderson Cancer Center, Vall d’Hebron University Hospital, Hospital Germans Trias i Pujol, and Parc Taulí Universitary Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.