Article Text

Abstract
Background:The acute respiratory distress syndrome (ARDS) is characterized by protein-rich oedema in the alveolar spaces, a feature in which Fas-mediated apoptosis of the alveolar epithelium has been involved.
Objective:To determine whether Fas activation increases protein permeability by mechanisms involving disruption of the paracellular tight junction (TJ) proteins in the pulmonary alveoli.
Methods: Protein permeability and the expression of TJ proteins were assessed in vivo in wild-type and Fas-deficient lpr mice 16 hours after the intratracheal instillation of recombinant human soluble Fas ligand (rh-sFasL), and at different time points in vitro in human pulmonary alveolar epithelial cells (HPAEpiC) exposed to rh-sFasL
Results:Activation of the Fas pathway increased protein permeability in mouse lungs and altered the expression of the TJ proteins occludin and zonula occludens-1 in the alveolar–capillary membrane in vivo and in human alveolar epithelial cell monolayers in vitro. Blockade of caspase-3, but not inhibition of tyrosine kinase dependent pathways, prevented the alterations in TJ protein expression and permeability induced by the Fas/FasL system in human alveolar cell monolayers in vitro. We also observed that both the Fas-induced increase of protein permeability and disruption of TJ proteins occurred before cell death could be detected in the cell monolayers in vitro.
Conclusion:Targeting caspase pathways could prevent the disruption of TJs and reduce the formation of lung oedema in the early stages of ARDS.
- pulmonary oedema
- innate immunity
- ards
Statistics from Altmetric.com
Key messages
What is the key question?
The key question of our study is whether the alteration of tight junction proteins in the alveolar epithelium is a mechanism by which the activation of the Fas/FasL system increases pulmonary permeability in acute lung injury, even in the absence of apoptotic cell death.
What is the bottom line?
The activation of the Fas/FasL system is capable of increasing protein permeability of the alveolar epithelium in vivo and in vitro by mechanisms involving caspase-3-dependent alterations of tight junction proteins (occludin and zonula occludens (ZO)-1) prior to and/or independently of the development of cell death.
Why read on?
This is the first study that links Fas activation with the alterations of tight junction proteins in the pulmonary alveolar epithelium in acute lung injury, suggesting that targeting this pathway may be a suitable strategy to preserve the alveolar epithelial function and to reduce lung oedema in acute lung injury.
Introduction
Accumulation of protein-rich oedema fluid into the alveoli and interstitium occurs in the early stage of the acute respiratory distress syndrome (ARDS), due to an increase in protein permeability of the alveolar–capillary membrane and to an impairment of the alveolar fluid clearance capability of the alveolar epithelium.1 The alveolar epithelial barrier is considered a major player in the formation and resolution of this oedema.2 3 The permeability and the alveolar fluid clearance capability of the alveolar epithelium are regulated by tight junctions (TJs), which are intercellular structures that prevent the passage of molecules through the paracellular spaces.4–6 Also, they maintain cellular polarity and regulate transcellular transport and a variety of intracellular signals.7 8 They are composed of transmembrane proteins such as occludins, claudins, tricellulin and other junction adhesion molecules (JAM), as well as cytoplasmic proteins such as zonula occludens (ZO)-1, ZO-2 and ZO-3 that ultimately bind to the actin fibres of the cytoskeleton.4–6 Preclinical studies have shown that TJs can be directly altered by various insults such as mechanical stress, viral and bacterial pathogens or their products (eg, endotoxin).9–12 TJ proteins can also be altered by the activation of different cell signalling pathways, including caspases and protein kinases.13–18 However, the exact role of TJs on the disruption of the alveolar epithelial barrier has not been fully elucidated in ARDS.
Activation of the receptor protein Fas by its natural ligand, FasL, is considered to play an important role in the development of ARDS.19–23 Our previous studies showed that activation of the Fas/FasL system increased alveolar–capillary protein permeability and impaired alveolar fluid clearance in mice, leading to lung oedema formation, by mechanisms involving caspase-mediated apoptosis in the alveolar walls.21 In addition, activation of the Fas/FasL system induces inflammatory responses in the lung, including cytokine release from epithelial cells via activation of protein kinases.20 22 24 In most models of acute lung injury, however, the number of apoptotic cells in the alveolar walls was relatively small. Although this situation could be explained by a rapid clearance of apoptotic cells, it is conceivable that pulmonary oedema mediated by Fas/FasL may not be exclusively due to the ultimate death of alveolar epithelial cells. We hypothesised that Fas activation leads to lung oedema formation by altering the structure of TJs without requiring the ultimate death of the cells.
In the present study, we determined the effect of human soluble FasL on pulmonary TJ proteins in mouse lungs in vivo and in human primary alveolar epithelial cell monolayers in vitro. We found that Fas activation by human soluble FasL increased the permeability and altered the expression and distribution of the TJ proteins occludin and ZO-1 in alveolar epithelial cells. Such changes occurred prior to cell death and were initially mediated by the activation of the apoptotic precursor caspase-3 but not by proinflammatory tyrosine kinase pathways.
Material and methods
Reagents
The long form of recombinant human soluble FasL (rh-sFasL) from ENZO Life Science (San Diego, California, USA) was used for in vivo studies. For in vitro experiments, we used a long form of rh-sFasL (103–281 aa, containing the stalk region) and a short form (134–281 aa, not containing the stalk region) from Abcam, as indicated in each experiment. For immunoblotting, we used the following reagents: monoclonal mouse antioccludin (clone OC-3F10) (Invitrogen), polyclonal rabbit anti-ZO-1 (Invitrogen), Alexa Fluor 546 conjugated-goat antimouse IgG (H+L) antibody and Alexa Fluor 568 conjugated-goat antirabbit IgG (H+L) (ThermoFisher Scientific), 4′,6-diamino-2-fenilindol (DAPI) from Invitrogen. Proteinase K, protein block serum-free, fluorescent mounting medium and microscope slides were obtained from DAKO. In situ cell death detection kit-fluorescein for the terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) technique was purchased from Roche. The irreversible caspase-3 inhibitor zDEVD.FMK and the negative control for caspase inhibitors zFA.FMK were purchased from BD Pharmingen. The tyrosine kinase inhibitor genistein was obtained from Sigma-Aldrich. The universal tyrosine kinase assay kit was obtained from Takara-Clontech. Fluorescein (FITC)-human serum albumin protein (full length) was purchased from Abcam. Unless otherwise indicated, all other chemicals were purchased from Sigma-Aldrich.
Animal studies
Mice were housed in conventional cages with ad libitum access to tap water and standard chow, in humidity-controlled (50%) and temperature-controlled (22°C–23°C) rooms with 12 hours/12 hours light/dark cycles. In the morning, male C57BL/6 mice or naturally occurring mutant mice lacking functional Fas receptor (B6.MRL-Fas lpr/J mice) (lpr mice) (The Jackson Laboratory, Bar Harbor, Maine, USA) weighing 25–30 g were anaesthetised with inhaled 2%–5% isofluorane and randomised to treatment with one intratracheal instillation of rh-sFasL (25 ng/g) or phosphate-buffered saline (PBS). The PBS group was instilled first to avoid cross-contamination with soluble FasL (sFasL). The number of mice used per group was 10. For the histology analyses, five additional mice were included. The dose of rh-sFasL was chosen from a prior dose–response experiment showing that 25 ng/g of FasL was the minimal dose causing the highest level of neutrophil recruitment into the alveolar airspaces (data not shown). After the instillations, mice were allowed to recover from anaesthesia and returned to their cages with free access to food and water. The mice were euthanised 16 hours after instillation by an intraperitoneal injection of pentobarbital (120 mg/kg) and exsanguinated by closed cardiac puncture. The thorax was opened rapidly, the trachea was cannulated with a 20-gauge catheter, the left hilum was clamped and the left lung was removed, rapidly weighed on a precision balance and flash-frozen in liquid nitrogen for protein and enzyme analyses. The right lung was either fixed by intratracheal instillation of 4% paraformaldehyde at a transpulmonary pressure of 15 cm of water, and then embedded in paraffin for histology analysis or instilled with five separate 0.5 mL aliquots of 0.9% NaCl containing 0.6 mM EDTA at 37°C to obtain the bronchoalveolar lavage (BAL) fluid.
Analysis of mouse BAL fluid
The BAL fluid samples from the right lung were processed immediately for total and differential cell counts. Total white cell counts were performed with a haemocytometer, and differential counts were performed by using Advia 2120i System analyser. The remainder of the lavage fluid was spun at 200 g for 30 min, and the supernatant was removed aseptically and stored in individual aliquots at −80°C. The concentration of IgM in BAL fluid was measured by ELISA (Bethyl Laboratories, Montgomery, Texas, USA) following the manufacturer’s instructions. The lower limit of detection of the IgM assay was 20 ng/mL.
Histological methods in mouse lung tissue
Paraffin-embedded murine lung tissue sections (4 µm thick) were obtained. TUNEL fluorescent staining for detection of DNA damage in situ was performed according to the manufacturer’s instructions (Roche Diagnostics). Light and fluorescence microscopy were performed using a Nikon Eclipse 80i microscope. Measurement of TUNEL-positive cells was performed in a blinded manner on eight randomly generated visual fields at magnification of 200×. Double labelling fluorescence techniques were used to evaluate TUNEL-positive cells in relation with occludin or ZO-1 staining in the lung tissue sections. Briefly, paraffin-embedded sections were deparaffinised in xylene and rehydrated in 100%, 95% and 70% ethanol. Next, the sections were heated at 95°C for 20 min in antigen retrieval buffer with sodium citrate (0.29 g citrate+0.1% Triton in 100 mL ddH2O). Next, the sections were incubated for 30 min with proteinase K at 37°C and permeabilised with 0.3% Triton X-100/PBS (PBST). The TUNEL method was performed first (incubation for 1 hour with TUNEL reaction mixture at 37°C in the dark), followed by washes with PBS. The tissue sections were blocked with protein block serum-free solution (DAKO) for 30 min in the dark. For occludin or ZO-1 immunofluorescence staining, the sections were incubated for 1 hour with a mouse monoclonal antioccludin antibody (Clone OC-3F10) (1:100) or a rabbit polyclonal anti-ZO-1 antibody (1:50) in PBS overnight at 4°C in a moist chamber in the dark. After being washed in PBS, the sections were incubated for 1 hour in PBS containing Alexa Fluor 546-conjugated goat antimouse or Alexa Fluor 546-conjugated goat antirabbit (1:250), respectively, at room temperature in the dark. After washes in PBS, the sections were stained with DAPI (300 nM) and incubated for 5 min in the dark. The sections were then mounted with a fluorescent mounting medium and analysed by light and fluorescence microscopy by acquiring sets of serial optical sections in both the fluorescence and the differential interference contrast mode.
Measurements in lung homogenates
For caspase-3 activity measurements, 20 mg of lung tissue were homogenised in lysis buffer provided by the Caspase-3/CPP32 Fluorometric Assay Kit (Biovision), following manufacturer’s instructions. For occludin and ZO-1 protein measurements in mouse lungs, the membrane and cytosolic proteins from 20 mg of mouse lung tissue were separated and isolated using the Mem-PER Plus membrane protein extraction kit (Thermo Scientific). Protease and phosphatase inhibitors (Roche) were added to the permeabilisation and solubilisation buffers. Briefly, lung tissue was washed in a microcentrifuge tube with cell wash solution. The lung tissue was cut into small pieces and homogenised in sample tubes filled with ceramic beads and permeabilisation buffer using a MagNA lyser (Roche Life Science). To separate cytosolic and membrane fractions, lung homogenates were incubated in permeabilisation buffer for 10 min on ice and then centrifuged at 16 000×g for 15 min at 4°C. The supernatant containing cytosolic proteins was separated from the pellet. To obtain the membrane proteins, the pellet was resuspended in solubilisation buffer and incubated for 30 min at 4°C, and then centrifuged at 16 000×g for 15 min at 4°C. Aliquots of cytosolic and membrane protein fractions were stored at −80°C. Total protein concentrations of the membrane and cytosolic fractions were measured by the bicinchoninic acid method (Pierce BCA Protein assay, Rockford, Illinois, USA). Occludin and ZO-1 were measured in lung lysates by ELISA kits from Cloud Clone Corporation and Mybiosource, respectively. The sensitivities of the immunoassays were 0.132 ng/mL for occludin and 0.1 ng/mL for ZO-1. The fluorescence signal was measured using a fluorescence plate reader.
Cell culture
Human pulmonary alveolar epithelial cells (HPAEpiC) isolated by ScienCell Research Laboratories from human lung tissue were provided by Innoprot. HPAEpiC are composed of alveolar type I and type II epithelial cells, but type I cells are predominant, as the number of type II cells decreases with successive passages. HPAEpiC were grown in alveolar epithelial cell medium, which was buffered with HEPES and bicarbonate and supplemented with 2% heat-inactivated fetal bovine serum, 1% epithelial cell growth supplement and 1% penicillin/streptomycin solution (ScienCell Research Laboratories) at 37°C and 5% CO2. For the bioassay experiments and measurement of albumin permeability, the HPAEpiC were seeded in transparent collagen-coated transwell inserts with a membrane pore size of 0.4 µm and surface area of 0.33 or 1.12 cm2 (Corning, Lowell, MA) at a density of 1×104 cells/well or 2×104 cells/well, respectively. The culture medium was added to the upper and lower compartment of each transwell, and the cells were allowed to reach 100% confluence, which was confirmed by phase contrast light microscopy. Next, medium containing rh-sFasL at concentrations of 0 ng/mL , 33.3 ng/mL, 100 ng/mL, 300 ng/mL or 600 ng/mL (as specified in each experiment) or PBS was added to the upper and lower compartments. As indicated in each experiment, the cells were preincubated for 1.5 hours with the caspase-3 inhibitor zDEVD.fmk (10 µM), the negative control of the caspase-inhibitors zFA.fmk (10 µM), the tyrosine kinase inhibitor genistein (30 µM) or vehicle (dimethyl sulfoxide, DMSO). All these reagents were reconstituted in DMSO and diluted in medium to obtain their working concentrations on the cells, so the vehicle/control condition corresponds to pure DMSO diluted in medium in the same manner. After incubation for 2 hours with FasL, the culture medium was removed from the upper and lower transwell compartments, spun at 200 g and stored in individual aliquots at −20°C for cytokine measurements.
Measurement of FITC-albumin permeability in cultured alveolar epithelial cell monolayers
After 2 hours of incubation with human recombinant FasL (with or without preincubation with zDEVD.fmk, zFA.fmk or genistein), the permeability to FITC-albumin of the HPAEpiC monolayers was determined by the addition of medium supplemented with 0.05% FITC-albumin in the lower compartments. The fluorescence intensity was measured in the medium collected from the upper and the lower transwell compartments using a fluorescence plate reader. The permeability ratio to albumin was expressed as a percentage of top FITC-albumin fluorescence relative to bottom FITC-albumin fluorescence, which reflects the passage of FITC-albumin into the upper compartment in each transwell.
Cell viability and caspase-3 activity in alveolar epithelial cell monolayers
Cell viability was assessed using the PrestoBlue assay (Invitrogen), which incorporates an oxidation/reduction indicator that fluoresces red in the presence of active cellular metabolism. Data are shown as the percentage of cell death, which was calculated as followed: cell death (%)=100 × ([live cell fluorescence – experimental fluorescence]/live cell fluorescence). Untreated live cell fluorescence corresponds to the fluorescence of cells in medium only. Finally, the cells were lysed, and caspase-3 activity was measured in each well using a Caspase-3 Fluorometric Assay Kit (Biovision) and a fluorescence plate reader.
Protein and tyrosine kinase activity measurements in alveolar epithelial cell monolayers
Interleukin (IL)-8 and IL-6 in the culture medium of HPAEpiC monolayers incubated in different conditions, as indicated in each experiment, were measured by ELISA (R&D system) according to manufacturer’s instructions. The sensitivity of the ELISA kit for IL-8 was 7.5 pg/mL and for IL-6 was 9.38 pg/mL. Tyrosine kinase activity was measured in cell lysates by using a universal tyrosine kinase assay kit from Takara-Clontech according to manufacturer’s instructions. In additional cultured cells, the occludin and ZO-1 protein levels of the HPAEpiC monolayers were measured in cell lysates by ELISA kits from Cloud-Clone, following the manufacturer’s instructions. The sensitivity of the ELISA kits for occludin was 0.063 ng/mL and for ZO-1 was 0.059 ng/mL.
Immunocytochemistry and TUNEL technique
After treatment with human FasL or medium, the HPAEpiC seeded in transwell membranes were washed with prewarmed PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. The transwell membrane was gently cut and placed over a paraffin film with cells facing up. After three washes with PBS, the cells were permeabilised for 10 min with PBS containing 0.3% triton on ice. After washes with PBS, the cell monolayers were blocked with protein block serum-free (DAKO) for 30 min at room temperature. The cells were then incubated with a mouse monoclonal antioccludin (clone OC-3F10) or a rabbit polyclonal anti-ZO-1 antibody (1:200) overnight at 4°C in a moist atmosphere. Subsequently, the cell monolayers were incubated for 1.5 hours with Alexa Fluor 546-conjugated goat antimouse or Alexa Fluor 546-conjugated goat antirabbit secondary antibody (1:250) at room temperature in the dark, stained with DAPI (300 nM) and mounted with fluorescent mounting medium and analysed by fluorescence microscopy. For the experiments combining the TUNEL method (to label DNA fragments) and the immunocytochemistry (to label occludin or ZO-1 proteins), the cells were permeabilised as explained above, washed twice in PBS and incubated with TUNEL reaction mixture for 60 min at 37°C. After three washes in PBS, the same immunocytochemistry protocol was performed in the cells as explained above. The labels were visualised by fluorescence microscopy using a green wavelength (TUNEL), a red wavelength (occludin and ZO-1) and a blue wavelength (DAPI).
Statistical analysis
Quantitative variables were reported as mean (95% CI) unless otherwise indicated. Differences between three or more groups were analysed using one-way analysis of variance followed by the Bonferroni’s post hoc tests for variables with normal distribution or the Kruskal-Wallis test followed by the Dunn’s test for those without a normal distribution. Differences between two groups were analysed using the two-tailed unpaired Student’s t-test for variables with a normal distribution or the Mann-Whitney test for variables without a normal distribution. A logarithmic transformation (log10) was used to reduce the heterogeneity of variances when these were significantly different. The sample size was calculated in initial experiments that evaluated the changes in the concentration of occludin and ZO-1 in the lungs of wild-type mice treated with FasL versus PBS. We detected a 50% fall of the concentration of occludin and ZO-1 in FasL-treated mice compared with control mice with a maximal SD of 1.8. Considering a power (1-β) of 90% and a significance level (α) of 5%, the calculated number of mice needed was from 8 to 10 per group. A p value less than 0.05 was considered statistically significant. The statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software).
Results
Human FasL increased pulmonary protein permeability, neutrophil recruitment and lung weight in mice
Intratracheal instillation of rh-sFasL in wild-type mice resulted in increases of the concentration of IgM and of the number of polymorphonuclear (PMN) leukocytes and in a decrease of the number of macrophages in BAL fluid collected 16 hours after the instillation (figure 1A–C). These changes in wild-type mice were further accompanied by an increase of the left lung weight-to-total body weight ratio (figure 1D) compared with control mice treated with PBS. In contrast, rh-sFasL instillation did not induce changes in Fas-deficient lpr mice. Control wild-type and lpr mice instilled with PBS had similar IgM concentration and number of PMN and macrophages in the BAL fluid at baseline (figure 1A–C).
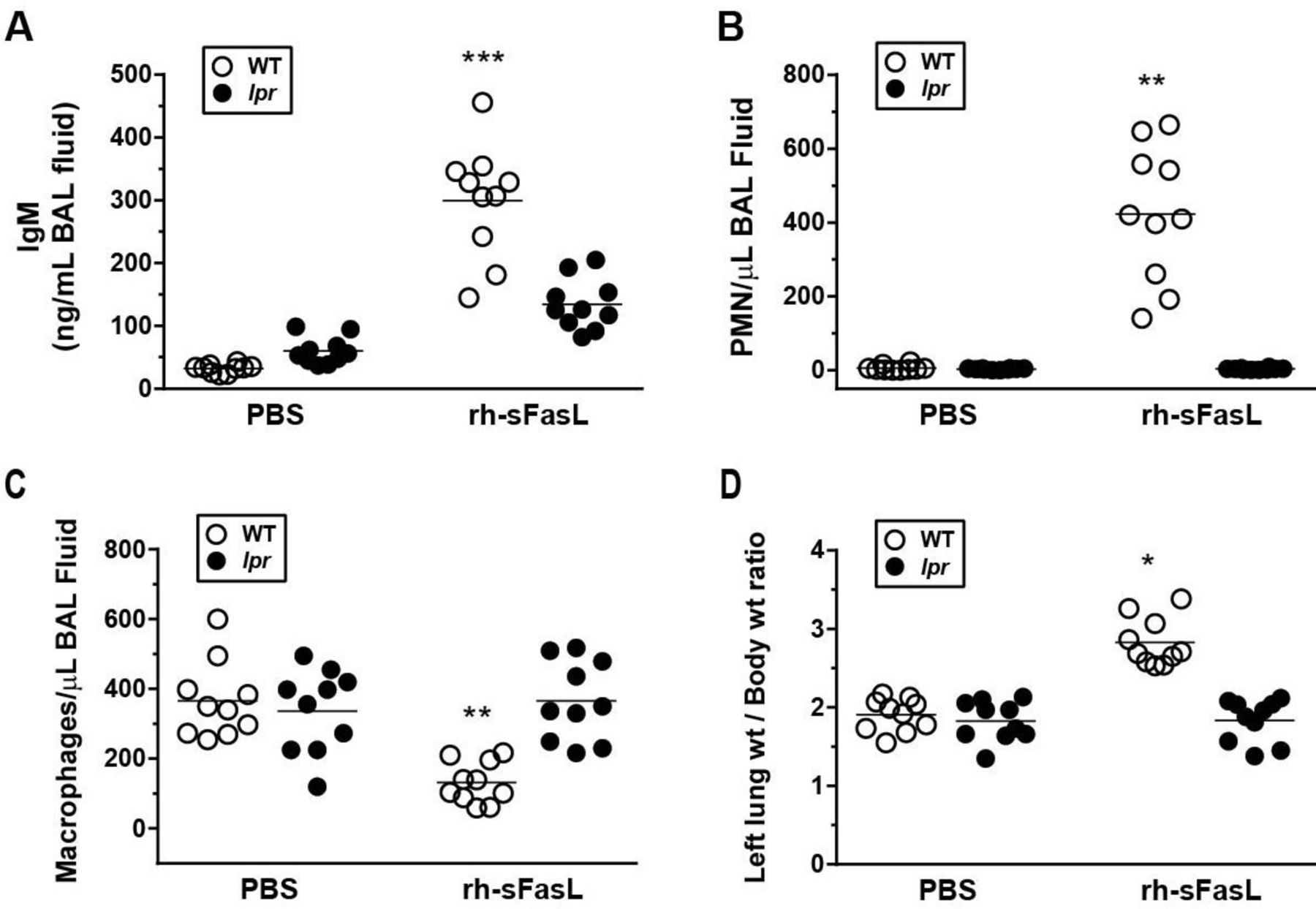
Effects of FasL on pulmonary protein permeability (A), cellular inflammatory responses (B and C) and left lung weight (D) in wild-type (WT) and Fas-deficient lpr mice in vivo. Mice were treated with intratracheal instillation of recombinant human soluble FasL (rh-sFasL; 25 ng/g body wt) or phosphate-buffered saline (PBS, as control), and then studied 16 hours later. The graphs show the effect of rh-sFasL on (A) the concentration of IgM (a plasma protein of large size, 900 kDa), (B) the number of polymorphonuclear (PMN) leukocytes and (C) the number of macrophages in bronchoalveolar lavage (BAL) fluid, and on (D) the left lung wt-to-body wt ratio. In the graphs, each dot represents an individual mouse (n=10 per group). Horizontal bars represent means. *p<0.005, **p<0.001, ***p<0.0001 versus WT-PBS group.
Human soluble FasL altered the expression of occludin and ZO-1 in mouse lungs in vivo
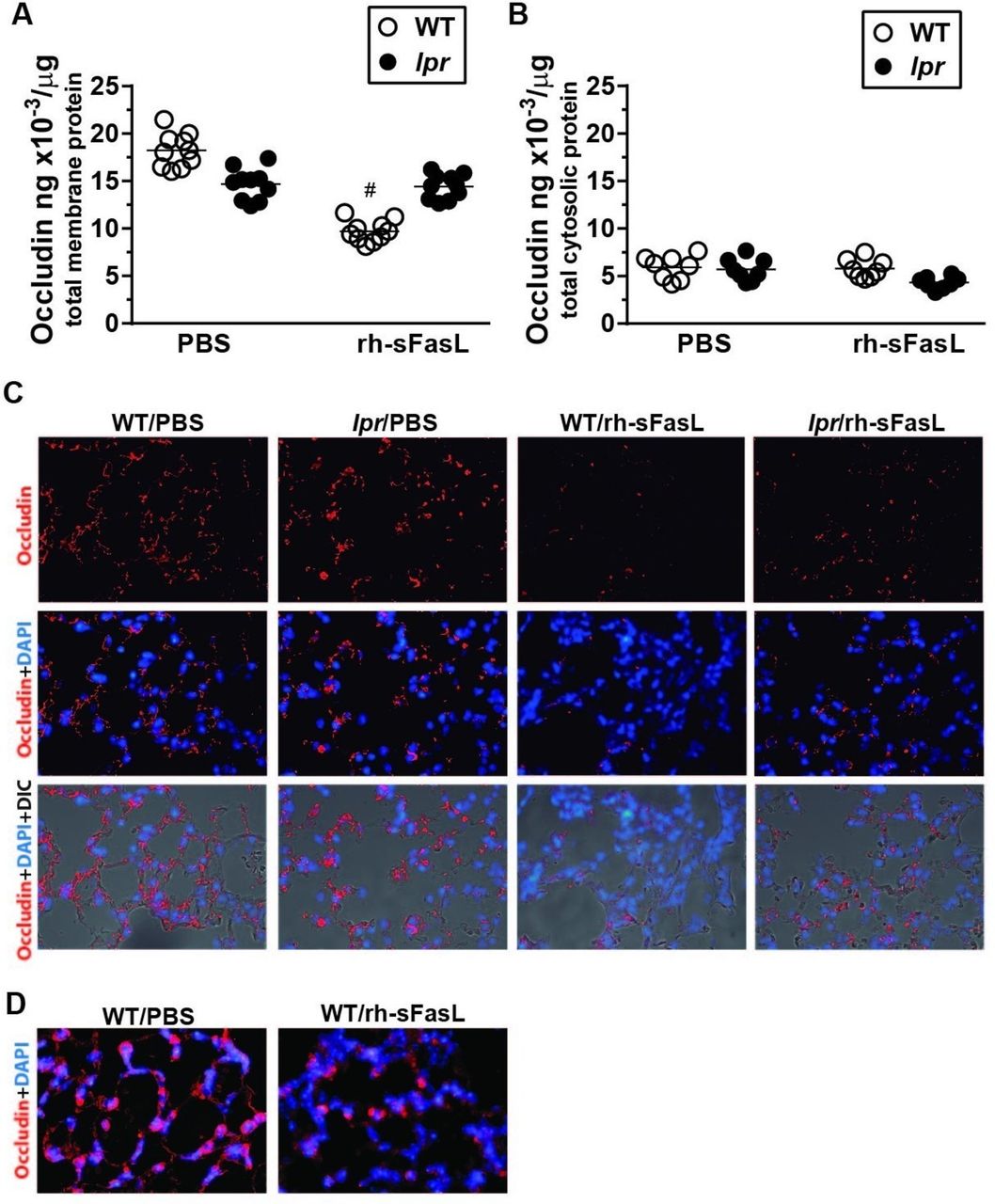
To determine whether the disruption of the TJ proteins occludin and ZO-1 was a potential mechanism responsible for the increase in protein permeability and lung oedema formation induced by FasL, we first measured the concentrations of occludin and ZO-1 in the membrane and cytosolic fractions of lung tissue extracts collected 16 hours after the intratracheal instillation of rh-sFasL or PBS in wild-type and lpr mice. As expected, the membrane fraction was enriched in occludin and ZO-1 proteins compared with the cytosolic fraction of the lung tissue extracts in all groups of mice (figure 2A,B and figure 3A,B). For both fractions, the concentrations of occludin and ZO-1 were similar in wild-type and lpr mice treated with PBS (figure 2A,B and figure 3A,B), despite a non-significant trend for a reduced expression of occludin in the membrane fraction in lpr mice (figure 2A). Whereas the instillation of rh-sFasL in wild-type mice decreased the protein concentration of occludin in the membrane fraction and of ZO-1 in both the membrane and cytosolic fractions, the instillation of rh-sFasL in lpr mice caused no changes in the protein levels of occludin or ZO-1 in any of the fractions (figure 2A,B and figure 3A,B).

Human soluble FasL alters the expression of occludin in the lungs of wild-type (WT) but not in the lungs of lpr Fas-deficient mice. Mice were treated with intratracheal instillation of recombinant human soluble FasL (rh-sFasL; 25 ng/g body wt), and then studied 16 hours later. As control, WT and lpr Fas-deficient (lpr) mice were treated with phosphate-buffered saline (PBS) via intratracheal instillation. We measured the concentration of occludin in the membrane and cytosolic fractions in mouse lungs by ELISA (A and B, respectively). The merged images of fluorescence microscopy and light microscopy with differential interference contrast (DIC) show occludin protein signal (red) and cell nuclei (DAPI staining, blue) over the structure of the alveolar walls. Original image magnification ×200 (C). Representative images at larger magnification (×400) show the decreased expression of occludin protein (red signal) in the lung of WT mice treated with rh-sFasL compared with mice treated with PBS (D). In the graphs, each dot represents an individual mouse (n=10 per group). Immunofluorescence analyses were performed in 5 mice per group. Horizontal bars represent means. #P<0.0001 versus WT-PBS group.

Human soluble FasL alters the expression of ZO-1 in the lungs of wild-type (WT) but not in the lungs of lpr Fas-deficient mice. WT and lpr Fas-deficient (lpr) mice were treated with intratracheal instillation of recombinant human soluble FasL (rh-sFasL; 25 ng/g body wt), and then studied 16 hours later. As control, WT and lpr mice were intratracheally instilled with phosphate-buffered saline (PBS). We measured the concentration of zonula occluden (ZO)-1 in the membrane and cytosolic fractions in mouse lungs by ELISA (A and B, respectively). The merged images of fluorescence microscopy and light microscopy with differential interference contrast (DIC) show occludin protein signal (red) and cell nuclei (DAPI staining, blue) over the structure of the alveolar walls. Original image magnification ×200 (C). Representative images at larger magnification (x400) show the decreased expression of ZO-1 protein (red signal) in the lung of WT mice treated with rh-sFasL compared with mice treated with PBS (D). In the graphs, each dot represents an individual mouse (n=10 per group). Immunofluorescence analyses were performed in 5 mice per group. Horizontal bars represent means. *P<0.005 versus WT-PBS group.
Immunofluorescence techniques in lung tissue sections showed that occludin and ZO-1 were homogenously distributed in the cells of the alveolar walls in wild-type and lpr mice treated with PBS as well as in lpr mice treated with rh-sFasL. In contrast, the lungs of wild-type mice treated with FasL showed areas with loss of occludin and ZO-1 expressions in the alveolar walls (figure 2C,D and figure 3C,D).
Altogether, these data showed that intratracheal instillation of rh-sFasL caused changes in occludin and ZO-1 proteins in the lung of wild-type mice, but not in Fas-deficient lpr mice, suggesting that human soluble FasL alters the expression and normal distribution of these TJ proteins through activation of Fas receptor in mouse lungs in vivo.
FasL-induced apoptosis is locally associated with altered expression of occludin and ZO-1 in mouse lungs in vivo
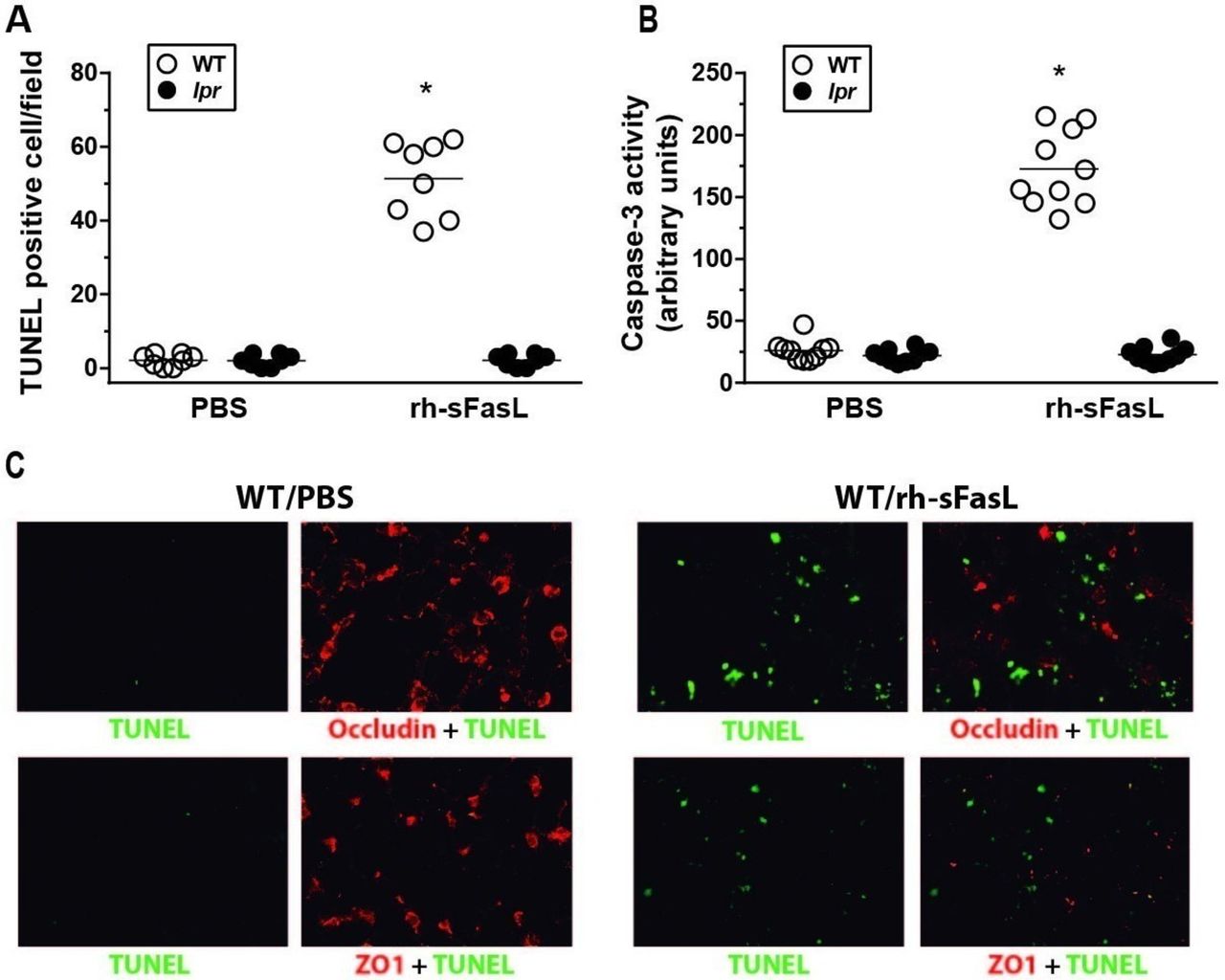
Compared with PBS treatment, instillation of rh-sFasL significantly elevated the number of cells with nuclei containing DNA strand breaks (TUNEL-positive signal) and caspase-3 activity in the lungs of wild-type mice (figure 4A,B, respectively). Importantly, the merge of immunofluorescence images from FasL-treated wild-type mice showed that occludin and ZO-1 protein signals were diminished in the alveolar walls only in areas with TUNEL positive cells (figure 4C). In contrast, no increase of caspase-3 activity or TUNEL positive cells were observed in lpr mice. These results suggest an association between FasL-induced apoptosis and the disruption of TJ proteins in mouse lungs in vivo.

Fas-induced apoptosis is locally associated with altered expression of occludin and ZO-1 in mouse lungs in vivo. Wild-type (WT) and Fas deficient (lpr) mice were treated with one intratracheal instillation of recombinant human soluble FasL (rh-sFasL; 25 ng/g body wt) or phosphate-buffered saline (PBS, as control). At 16 hours postinstillation, we measured (A) the number of nuclei containing DNA strand breaks (TUNEL-positive signal) in lung tissue sections and (B) caspase-3 activity in the lung homogenates. Merged images of representative lung tissue sections from PBS or FasL-treated WT mice (C) showed that occludin and ZO-1 protein signals (red) were diminished in the alveolar walls only in areas with TUNEL-positive cells (green). Original image magnification ×400. Immunofluorescence analyses were performed in 5 mice per group. In the graphs, each dot represents an individual mouse (n=10 mice per group). Horizontal bars represent means. *P<0.01 versus WT-PBS group. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.
Human FasL increases alveolar epithelial cell permeability prior to cell death
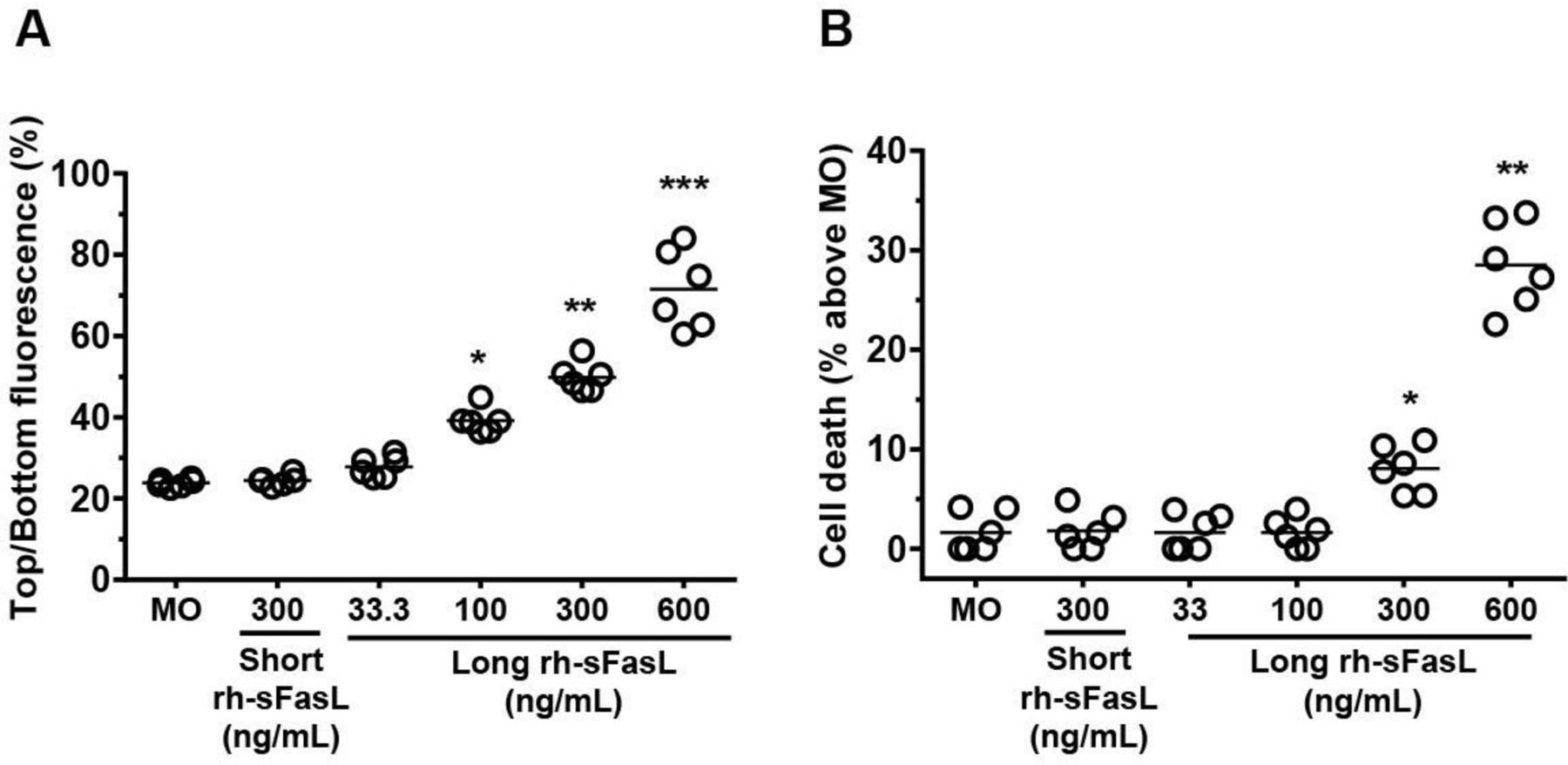
To determine whether cell death by apoptosis was a requirement for the increase of alveolar permeability induced by FasL or there were other apoptosis-related events contributing to this alteration, we measured the protein permeability and cell death in HPAEpiC monolayers after 2 hours of incubation with increasing concentrations (from 33 ng/mL to 600 ng/mL) of a cytotoxic long form of human recombinant sFasL (figure 5A,B). Cells incubated with medium only or with a short form of human recombinant soluble FasL (300 ng/mL), which is not cytotoxic for epithelial cells,20 were used as controls. As expected, HPAEpiC expressed aquaporin-5, a water channel protein specifically expressed in alveolar type I cells in the lung, and showed the characteristic staining pattern of actin (online supplementary figure 1). Incubation of the HPAEpiC monolayers with 33 ng/mL, 100 ng/mL, 300 ng/mL and 600 ng/mL of the long form of FasL resulted in dose-dependent increases of FITC-albumin permeability (28.4% (95% CI 26.35 to 30.5%) (p=0.3703), 39.1% (95% CI 36.5% to 41.1%) (p=0.0120), 49.9% (95% CI 47.4% to 52.7%) (p=0.0001) and 71.5% (95% CI 64.4% to 80.5%) (p<0.0001), respectively), compared with medium only (23.8% (95% CI 23.1% to 24.6%)) (figure 5A). The early alteration of transepithelial permeability by FasL was also observed when using the Transepithelial Electrical Resistance method (see online supplementary figure 2). Importantly, the percentage of cell death at the 2-hour time point was only increased by the 300 ng/mL and 600 ng/mL doses of the long form of human sFasL (% cell death: 8.0% (95% CI 5.5% to 10.5%) (p=0.022) and 25.2% (95% CI 22.5% to 27.9%) (p=0.0025), respectively), compared with non-treated cells (figure 5B). Although all doses of sFasL resulted in apoptosis with long (>6 hours) incubation times (see online supplementary figure 3), the 100 ng/mL dose of long sFasL did not cause detectable cell death despite enhancing protein permeability at the 2-hour time point. Exposure to the non-cytotoxic short form of rh-sFasL (300 ng/mL) did not induce cell death nor changed the protein permeability of the alveolar epithelial cell monolayers (figure 5A,B). These data suggested that Fas signalling contributes to the loss of the alveolar epithelial barrier function by mechanisms that precede the ultimate death of the cells.
Supplementary file 1

Human soluble FasL increases alveolar epithelial cell permeability prior to cell death in vitro. The permeability to FITC-albumin (A) and the percentage of cell death (B) were measured in human pulmonary alveolar epithelial cell (HPAEpiC) monolayers after 2 hours of incubation with increasing concentrations of a cytotoxic long form of human recombinant sFasL from 33 ng/mL to 600 ng/mL. Cells with medium only (MO) or with a non-cytotoxic short form of human recombinant soluble FasL (300 ng/mL) were used as controls. Results from three separate experiments performed in duplicate. Each dot of the graphs represents a single data point. Horizontal bars represent means. **P<0.05, **p<0.005, ***p<0.0001 versus MO.
The increase in protein permeability induced by soluble FasL is dependent on caspase-3 but not on tyrosine-kinase signalling-dependent cytokine production
To determine the signalling pathways by which human soluble FasL altered the lung epithelial barrier function, we performed experiments in HPAEpiC monolayers using the caspase-3 inhibitor zDEVD.fmk and the tyrosine-kinase specific inhibitor genistein.
First, we determined the minimal dose of zDEVD.fmk that blocked caspase-3 activity in HPAEpiC by preincubating them for 1.5 hours with different doses (from 1 µM to 100 µM) of zDEVD.fmk, its negative control peptide zFA.fmk or vehicle (DMSO). Then, the cells were exposed for 2 hours to medium only or to the two doses of rh-sFasL that increased protein permeability associated with no cell death (100 ng/mL) or only with minimal cell death (300 ng/mL). Compared with medium only, both doses of FasL increased the activity of caspase-3 in a dose-dependent manner in cells preincubated with vehicle (medium only: 30.9 (95% CI 26.0 to 35.8) arb. units vs FasL-100 ng/mL: 85.7 (95% CI 56.5 to 114.9) arb. units (p=0.0002) vs FasL-300 ng/mL: 273.6 (95% CI 242.1 to 305.1) arb. units (p<0.0001)) or with zFA.fmk (figure 6A). In contrast, preincubation with zDEVD.fmk at the dose of 10 µM totally prevented the increase in caspase-3 activity caused by both doses of FasL (figure 6A). Remarkably, preincubation with zDEVD.fmk, but not with vehicle or zFA.fmk, also abrogated the increase in FITC-albumin permeability caused by both doses of FasL (figure 6C). Whereas the dose of 100 ng/mL of FasL did not cause cell death in any of the groups as measured by PrestoBlue reagent, the small percentage of cell death caused by the 300 ng/mL dose of FasL was also prevented in cells preincubated with zDEVD.fmk (figure 6B). Preincubation with vehicle, zFA.fmk or zDEVD.fmk in cells not exposed to FasL did not alter caspase-3 activity, cell viability or protein permeability (figure 6A–C). These data showed that FasL increased protein permeability of HPAEpiC monolayers via a caspase-3-dependent pathway before cell death occurred.

Human sFasL increases protein permeability in human pulmonary alveolar epithelial cell (HPAEpiC) monolayers by mechanisms dependent on caspase-3, but not on tyrosine-kinase signalling-dependent cytokine production. HPAEpiC monolayers were preincubated with caspase-3 inhibitor zDEVD.fmk (or zFA.fmk or vehicle as controls) and exposed for 2 hours to rh-sFasL (100 ng/mL or 300 ng/mL) or medium only (MO) (A–C). After treatment, we measured: (A) caspase 3-activity, (B) percentage of cell death and (C) FITC-albumin permeability in these cell monolayers. HPAEpiC monolayers were also preincubated with the tyrosine-kinase-specific inhibitor genistein (or vehicle as control) (D–H) and exposed for 2 hours to recombinant human soluble FasL (rh-sFasL) (100 ng/mL or 300 ng/mL) or MO. After treatment, we measured interleukin (IL)-8 concentration in cell supernatant by ELISA (D), percentage of cell death by PrestoBlue (E), permeability to FITC-albumin (F), IL-6 concentration in cell supernatant by ELISA (G) and tyrosine kinase activity in the cell monolayers (H). Cell monolayers treated with medium only were used to determine 100% survival. Results from four separate experiments performed in duplicate. Each dot of the graphs represents a single data point. Horizontal bars represent means. *P<0.05, **p<0.001, ***p<0.0001 versus all conditions with MO; #p<0.05.
Because it is known that Fas activation in lung epithelial cells increases cytokine production simultaneously to the induction of apoptosis,22 we investigated if cytokine production was involved in the sFasL-induced alteration of the lung epithelial barrier function. First, we confirmed that the production of IL-8 by HPAEpiC monolayers was increased as soon as 2 hours after exposure to rh-sFasL at the doses of 100 ng/mL and 300 ng/mL compared with medium only (FasL 100 ng/mL: 9.28 pg/mL (95% CI 8.54 pg/mL to 10.02 pg/mL) (p<0.0001), FasL 300 ng/mL: 10.58 pg/mL (95% CI 9.58 pg/mL to 11.58 pg/mL) (p<0.0001) versus medium only: 5.28 pg/mL (95% CI 4.71 pg/mL to 5.84 pg/mL)). In the following experiments, HPAEpiC monolayers were preincubated with different concentrations (from 10 µM to 100 µM) of genistein or vehicle for 1.5 hours before exposure to rh-sFasL. The minimal dose of genistein that effectively prevented the increase in IL-8 expression induced by rh-sFasL without causing cell death was 30 µM (online supplementary figure 4). Preincubation with this dose of genistein (30 µM) significantly diminished tyrosine kinase activity in HPAEpiC treated for 2 hours with rh-sFasL (100 ng/mL or 300 ng/mL) or medium only (figure 6H). This was associated with a significant decrease in the expression of IL-8 and IL-6 induced by FasL in these cells (figure 6D,G). Despite blocking cytokine production and tyrosine kinase activity, preincubation with genistein did not prevent or attenuate the increased protein permeability induced by the two doses of rh-sFasL (100 ng/mL or 300 ng/mL) in human alveolar cell monolayers (figure 6F). Preincubation with 30 µM genistein did not cause nor influence sFasL-induced cell death either (figure 6E). These results suggested that tyrosine kinase activity and cytokine production were not required for the early increase in protein permeability induced by sFasL in alveolar epithelial cell monolayers.
Human sFasL alters the levels and distribution of occludin and ZO-1 proteins by caspase-3-dependent, tyrosine kynase-independent mechanisms
To determine whether the increase of protein permeability induced by sFasL in HPAEpiC monolayers was associated to alterations of TJ proteins, we assessed protein permeability and the concentration and distribution of occludin and ZO-1 in HPAEpiC monolayers 2 hours after exposure to rh-sFasL (100 ng/mL) or medium only. Compared with control cells, the exposure to rh-sFasL decreased the concentrations of occludin and ZO-1 in cell protein extracts, as measured by ELISA (figure 7A,B). Immunocytochemical evaluation of these HPAEpiC, predominantly type I cells, showed occludin signal mainly distributed along the cytoplasmic extensions of the cells but not exclusively at the membrane sites of cell–cell contacts as occurs in cuboidal epithelial cells. ZO-1 signal was mainly found at the nuclei and distributed along the cytoplasmic extensions and at sites of cell–cell contact. Exposure to rh-sFasL resulted in a global decrease of occludin and ZO-1 fluorescence that was particularly profound in the cytoplasmic extensions, while some signal remained close to the cell nuclei (figure 7C,D). Preincubation with zDEVD.fmk effectively prevented the sFasL-induced decrease of occludin and ZO-1 proteins evaluated both by ELISA (figure 7A,B) and immunocytochemistry (figure 7C). In contrast, preincubation with genistein or its vehicle (DMSO) or with zFA.fmk had no protective effects (figure 7A-C). Of note, preincubation with vehicle, genistein, zFA.fmk or zDEVD.fmk alone did not alter the expression of these TJ proteins (data not shown).

Human sFasL alters the levels and distribution of occludin and zonula occludens (ZO)-1 proteins in human pulmonary alveolar epithelial cells (HPAEpiC) in vitro by mechanisms dependent on caspase-3 but independent of tyrosine kinase-mediated cytokine production. After 2-hour exposure of recombinant human soluble FasL (rh-sFasL) (+FasL) at the dose of 100 ng/mL or medium only (−FasL), the concentrations of occludin and ZO-1 proteins were measured by ELISA in HPAEpiC monolayers preincubated with the caspase-3 inhibitor zDEVD.fmk, its inactive analogue (zFA.fmk) or the tyrosine kinase inhibitor genistein or vehicle (DMSO). Compared with control cells, rh-sFasL decreased the concentrations of occludin and ZO-1 in cell protein extracts (A and B, respectively). (C) Representative immunofluorescence images (original image magnification ×400) show the expression of occludin and ZO-1 proteins (red signals) localised along the cytoplasmic membrane of control cells in HPAEpiC monolayers (−rhFasL/+vehicle). Exposure to rh-sFasL resulted in a global decrease of occludin and ZO-1 fluorescence signals that was particularly profound in the cytoplasmic extensions, while some signal remained close to the cell nuclei (DAPI staining, blue signal). Only preincubation with the caspase-3 inhibitor zDEVD.fmk prevented the sFasL-induced decrease of occludin and ZO-1 proteins evaluated both by ELISA (A and B) and by immunocytochemistry (C). Representative immunofluorescence images at larger magnification (×1000) show the decreased expression of occludin and ZO-1 proteins (red signals) in HPAEpiC monolayers treated with rh-sFasL (+rh-sFasL/+vehicle) compared with control cells (−rh-sFasL/+vehicle) (D). Results from four separate experiments performed in duplicate. Each dot of the graphs represents a single data point. Horizontal bars represent means. *P<0.05, **p<0.01, ***p<0.001 versus their corresponding ¨–rh-sFasL¨ conditions.
Finally, to determine whether the loss of occludin and ZO-1 proteins induced by sFasL was an early event in the process of apoptosis, we incubated HPAEpiC monolayers with rh-sFasL (100 ng/mL) for 1, 2, 3 or 5 hours. By immunocytochemistry, we observed that the fluorescence signal of both occludin and ZO-1 proteins began to fall at least 2 hours before DNA fragmentation occurred in the nuclei of those cells (figure 8A,B).

FasL-mediated changes in the levels and distribution of occludin and zonula occludens (ZO)-1 proteins occur before nuclear DNA fragmentation in human pulmonary alveolar epithelial cells (HPAEpiC) in vitro. Representative fluorescence images of HPAEpiC monolayers exposed to recombinant human soluble FasL (rh-sFasL) (100 ng/mL) or medium only (MO) for 1, 2, 3 or 5 hours. The cells were labelled with the TUNEL method for the detection of DNA fragmentation (green signal), followed by labelling with an antibody to occludin or ZO-1 proteins (red signal). The merged image shows a decrease of occludin and ZO-1 fluorescence signals after exposure to FasL in a time-dependent manner. Despite this loss of occludin and ZO-1 fluorescence signal, no TUNEL-positive signal was observed during the first 3 hours of FasL exposure. However, TUNEL-positive cells were observed at 5-hour exposure of FasL. Original image magnification ×400. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling.
Discussion
The main goal of the present study was to determine whether Fas activation alters the alveolar epithelial function by mechanisms involving disruption of TJs in the lung. Our results demonstrated that activation of the Fas/FasL system altered TJ proteins and increased protein permeability of the alveolar–capillary membrane in mouse lungs in vivo. In alveolar epithelial cell monolayers, FasL caused a rapid alteration of TJ proteins associated with a marked increase in protein permeability prior to cell death by mechanisms dependent on caspase-3 activity. These data indicate that the Fas/FasL system increases pulmonary protein permeability by a direct effect on the alveolar epithelium that involves the alteration of its TJ proteins and permeability properties.
Our previous studies suggested that Fas-mediated apoptosis in alveolar epithelial cells impairs alveolar fluid clearance and increases pulmonary permeability by caspase-dependent mechanisms, leading to lung oedema formation in mouse lungs in vivo.21 This occurred in spite of a relatively small number of apoptotic cells observed in most animal models of acute lung injury involving the Fas/FasL system, such as those induced by endotoxin, bleomycin or mechanical ventilation.25 Although this situation could be explained by a rapid clearance of apoptotic cells, it is conceivable that the activation of apoptotic pathways also causes cellular changes that may contribute to lung oedema by mechanisms that do not depend on the ultimate death of epithelial cells in the lung. Reduced expression of TJ proteins has been identified in these animal models of acute lung injury together with an important dysfunction of the alveolar–capillary membrane.9 10 26 27 Those observations led us to explore whether the disruption of TJ proteins can be a mechanism by which Fas/FasL leads to lung oedema formation in acute lung injury, besides the loss of cells mediated by apoptosis.
In the present study, activation of the Fas/FasL system decreased the expression of two TJ proteins, occludin and ZO-1, in the alveolar walls of mouse lungs in vivo, which was associated with a significant increase in protein permeability and lung oedema formation, whereas mice lacking functional Fas receptor (lpr mice) were protected. Changes in the distribution of these TJ proteins occurred only in wild-type mice after FasL instillation with a tendency to form aggregates in the alveolar walls. Whereas the loss of ZO-1 protein results in a significant increase in epithelial protein permeability in most studies, a decreased expression of occludin is not always required to alter the barrier function in several non-pulmonary epitheliums.28 In line with this, the present study showed that the lower levels of occludin observed in lpr mice compared with wild-type mice was not associated with alveolar barrier dysfunction at baseline, which may be explained by their similar levels of ZO-1 proteins.
In human alveolar epithelial cells, a 2-hour exposure to human sFasL increased protein permeability of the alveolar epithelial monolayers together with a decrease in occludin and ZO-1 protein expression. These events occurred along with a prompt activation of the proapoptotic casaspe-3 enzyme, but the cells were still alive, and no cells were detached from the matrix after 2-hour treatment with sFasL. In addition, the morphology of these cells and the cell–cell contacts were similar to the untreated cells, except for a slight nuclear condensation. This is similar to other studies in which induction of apoptosis by other stimuli such as UV irradiation, staurosporine or anoxia resulted in reduced expression of occludin and/or ZO-1 proteins in non-pulmonary epithelial or endothelial cell lines in vitro as early as 6 hours, 3 hours and 30 min, respectively.13 29 30 These observations also occurred while the cells acquired a more rounded shape, but they were still attached, and no cell death was detectable by the cell viability assay (Prestoblue). In addition, we observed in a time-course experiment with HPAEpiC monolayers that exposure to FasL diminished that expression of occludin and ZO-1 at least 2 hours before nuclear DNA fragmentation (TUNEL positive) occurred. Also, cells showing important reduction of occludin and ZO-1 fluorescent signal but without DNA fragmentation (TUNEL negative) were detected. All these data indicated that disruption of TJ proteins was an early event of FasL-mediated apoptosis that occurred prior to nuclear DNA fragmentation. Importantly, this prompt disruption of TJ protein induced by FasL was capable of increasing protein permeability of the alveolar epithelium before complete cell death was established and therefore while the cells were still alive (figure 9). We cannot discard, however, that the ultimate death of cells can contribute to a further increase in protein permeability of the alveolar epithelial cell monolayers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic summarising the main findings of the study. (A) Effect of the instillation of recombinant human soluble FasL (rh-sFasL) into the lungs of wild-type C57Bl6 mice and lpr Fas-deficient mice at 16 hours postinstillation. (B) Effect of the 2-hours exposure to rh-sFasL in human pulmonary alveolar epithelial cells (HPAEpiC) alone or in combination with genistein or with zDEVD.fmk. TJ, tight junction; ZO, zonula occludens.
In our study, blockade of caspase-3 by using zDEVD.fmk prevented the deleterious effect of human FasL on TJ protein expression and on protein permeability in HPAEpiC monolayers in vitro. This was in agreement with our previous studies in which broad blockade of caspases significantly attenuated the increase in protein permeability and decreased apoptosis induced by human sFasL in mouse lung, whereas some cytokines such as IL-1β, IL-6 and KC were even increased.21 There is also evidence that TJ proteins, such as occludin, ZO-1 and ZO-2 are substrates of caspase-3 and other caspases after different proapoptotic stimuli, including Fas activation, in non-pulmonary apoptotic epithelial cells.13 14 31 Also, occludin has been found in the death-inducing signalling complex, where it colocalised with Fas, Fas-associated protein with death domain, active caspase-8 and caspase-3.31 32 This may explain the relationship found in our study between caspase-3 activation and disruption of TJ proteins after Fas/FasL-mediated apoptosis in alveolar epithelial cells (figure 9).
Activation of Fas via instillation of FasL induces apoptosis in mouse lungs and activates inflammatory responses with neutrophil recruitment to the alveolar spaces and increase in cytokine production in the lung.20 Instillation of FasL into the lung also reduced the number of macrophages in the BAL fluid, compared with PBS treated mice. This could be explained by a potential Fas-mediated activation of macrophages in the airspaces, because activated macrophages have been shown to adhere and interact with the epithelium, hindering their extraction by the BAL. Importantly, the Fas/FasL system has been shown to promote inflammatory responses and contribute to lung injury in different animal models of acute lung injury caused by lipopolysaccharide, sepsis or mechanical ventilation in which lpr-Fas-deficient mice and gld-FasL-deficient mice were protected.33 34 Isolated mouse alveolar epithelium and human distal airway epithelial cells exposed to FasL or to the Fas-activating Jo2 antibody undergo apoptosis through caspase-3-dependent pathways, along with an increase in KC/IL-8 release by mechanisms involving a rapid phosphorylation of several kinases, such as mitogen-activated protein kinases (MAPK), extracellular signal regulated kinases (ERK) and c-Jun N-terminal kinase (JNK). This rapid kinase phosphorylation was detected as early as 30 min after Fas activation in vitro.22 Also, it has been shown that Fas receptor physically interacts with tyrosine kinases, leading to a rapid cytokine production in non-pulmonary epithelial cells on Fas activation.35 36 Interestingly, it has been shown that occludin also binds to a complex set of tyrosine kinases.37 38 All these data point out a relationship between Fas, tyrosine kinases, cytokine production and occludin protein. By using genistein, a tyrosine kinase inhibitor, we diminished the activity of tyrosine kinase and blocked the early expression of IL-8, but it did not prevent the loss of TJ proteins—occludin or ZO-1—or the enhanced protein permeability caused by human sFasL in HPAEpiC monolayers. These observations suggest that the expression of cytokines mediated by tyrosine kinases does not play a major role in the initial disruption of TJ proteins and loss of barrier function of the alveolar epithelium caused by human FasL (figure 9). Because tyrosine kinase activation may also be involved in Fas-mediated apoptosis, we cannot discard that tyrosine kinases contribute to Fas-mediated alteration of TJ and epithelial permeability at later time-points.
The irreversible inhibitor of caspase-3 zDEVD.fmk used in this study can also have some inhibitory activity on other caspases involved in the apoptotic pathway, such as caspase-6, caspase-7, caspase-8 and caspase-10. In contrast, it cannot inhibit any of the caspases involved in cytokine activation (caspase-1, caspase-4, -caspase-5 and caspase-13). Therefore, we may assume that the effects of zDEVD.fmk were derived from the specific blockade of the caspase-dependent apoptotic pathway. Genistein is a highly specific inhibitor of protein tyrosine kinase by preventing tyrosine phosphorylation. To reduce unwanted potential effects of genistein on cell death, we used the minimal dose of genistein that prevented the increase of IL-8 induced by FasL without causing cell death (30 µM) (online supplementary figure 4). Finally, the doses of sFasL used in our study were similar to the concentration of sFasL found in the alveoli of patients with ARDS, which could range from 15 ng/mL to 150 ng/mL based on previous studies.23
Our study has some potential limitations. First, we evaluated mouse lungs in vivo and human alveolar epithelial cells in vitro, so caution must be exercised when extrapolating these findings to human lungs. Second, it is known that the extracellular matrix influences the expression of TJ proteins in the epithelium. Although we used unmodified HPAEpiCs, the response to FasL may differ between cells seated on collagen-coated membranes and those seated in the more complex basement membrane of human lungs. In addition, we did not assess other factors such as oxidative stress or mitochondrial damage that could also contribute to alveolar damage. Third, we have focused on alveolar epithelial cells, but Fas activation might also influence the expression of TJ proteins in the endothelium. Finally, we cannot discard that apoptosis and inflammation also influence the expression and function of TJ proteins at time points later than those evaluated in our study.
In conclusion, activation of proapoptotic caspase-3-dependent pathways of the Fas/FasL system seems an important mechanism responsible for the alteration of TJs—occludin and ZO-1—in the alveolar–capillary membrane that contributes to lung oedema formation. The disruption of TJs in the alveolar epithelium caused by Fas/FasL resulted in increased permeability long before the epithelial cells died by apoptosis (figure 9). These data provide a basis for further studies designed to determine whether the inhibition of the Fas/FasL pathway may prevent lung oedema formation and limit the progression of diffuse alveolar damage in ARDS, particularly at the earliest stages of the disease. Therapeutic strategies aimed to enhance proliferation and differentiation of the alveolar epithelial cells may be a better option at later stages when denudation of the alveolar epithelium is overtly present.
Acknowledgments
We would like to thank Mar Granados (pathology service of Hospital Universitario de Getafe) for her technical assistance with the immunofluorescence techniques.
References
Footnotes
RH and LP contributed equally.
Contributors Conception and design: RH. Acquisition, analysis and interpretation of data: all authors. Drafting and revising of the work: RH, LP, LM, GM-B and JAL. Final approval of the version: all authors.
Funding This work was supported by the Grants PI12/02451 and PI15/00482 (to RH) and PI15/01942 (to JAL) from the Instituto de Salud Carlos III, Ministerio de Economia y Competitividad, Madrid, Spain, and Fondos FEDER ’Una Manera de hacer Europa', and the Merit Award Number i01 BX002914 from the US Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service (to GM-B). LM is a recipient of a Miguel Servet Fellowship (CP12/03304) from the Instituto de Salud Carlos III (Spain), and RP was supported by a research initiation grant from CIBERES.
Competing interests None declared.
Patient consent Not required.
Ethics approval The animal protocol was approved by the Animal Care Committee of Hospital Universitario de Getafe (Madrid, Spain).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All the data from this work are freely available upon request.
Linked Articles
- Airwaves