Article Text

Abstract
Introduction Idiopathic pulmonary fibrosis (IPF) is characterised by accumulation of fibroblasts and myofibroblasts and deposition of extracellular matrix proteins. Sphingosine-1-phosphate (S1P) signalling plays a critical role in pulmonary fibrosis.
Methods S1P lyase (S1PL) expression in peripheral blood mononuclear cells (PBMCs) was correlated with pulmonary functions and overall survival; used a murine model to check the role of S1PL on the fibrogenesis and a cell culture system to study the effect of S1PL expression on transforming growth factor (TGF)-β- and S1P-induced fibroblast differentiation.
Results S1PL expression was upregulated in fibrotic lung tissues and primary lung fibroblasts isolated from patients with IPF and bleomycin-challenged mice. TGF-β increased the expression of S1PL in human lung fibroblasts via activation and binding of Smad3 transcription factor to Sgpl1 promoter. Overexpression of S1PL attenuated TGF-β-induced and S1P-induced differentiation of human lung fibroblasts through regulation of the expression of LC3 and beclin 1. Knockdown of S1PL (Sgpl1+/−) in mice augmented bleomycin-induced pulmonary fibrosis, and patients with IPF reduced Sgpl1 mRNA expression in PBMCs exhibited higher severity of fibrosis and lower survival rate.
Conclusion These studies suggest that S1PL is a novel endogenous suppressor of pulmonary fibrosis in human IPF and animal models.
- Idiopathic pulmonary fibrosis
Statistics from Altmetric.com
Video abstract
Key messages
What is the key question?
Is sphingosine-1-phosphate lyase (S1PL) an endogenous suppressor of pulmonary fibrosis?
What is the bottom line?
S1PL expression was elevated in lung tissues and lung fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) and bleomycin-challenged mice, and it reduced SGPL1 mRNA expression in peripheral blood mononuclear cells from patients with IPF correlated with higher severity of fibrosis and lower survival rate.
Why read on?
This is the first report that S1PL is an endogenous suppressor of pulmonary fibrosis through regulation of S1P signalling and autophagy in lung fibroblast, and the data presented here could be of potential significance towards therapeutic intervention in pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF), a chronic lung disease of unknown aetiology, is characterised by alveolar epithelial injury, and differentiation of epithelial cells and fibroblasts to myofibroblasts resulting in excessive collagen deposition, and lung scaring.1 Although molecular and cellular mechanisms of IPF are poorly defined, several factors such as oxidative stress, inflammation, coagulation and epithelial to mesenchymal transition have been implicated in the development and progression of IPF.2 ,3 As therapies for IPF are mostly ineffective, defining new mechanisms on IPF pathogenesis and development of novel therapeutic strategies are needed to manage the clinical pathology.
Sphingosine 1 phosphate (S1P), a naturally occurring bioactive sphingolipid, is generated in mammalian cells and tissues by phosphorylation of sphingosine catalysed by sphingosine kinase (SphK) 1 and 2.4 Lung tissues from patients with IPF and bleomycin-challenged and radiation-challenged mice exhibited enhanced expression of SphK1 and S1P/dihydro S1P levels, and genetic knockdown of SphK1, but not SphK2, increased survival and resistance to pulmonary fibrosis,5 suggesting that increased S1P generation mediated by SphK1 may be involved in the development and pathogenesis of pulmonary fibrosis. In fact, in bleomycin-induced murine model of lung inflammation and fibrosis, inhibition of SphK1 activity with a small molecular weight inhibitor-attenuated lung inflammation and fibrosis.5
S1P does not accumulate in cells/tissues under normal condition. Intracellularly generated S1P by SphKs is either secreted out via S1P transporter such as Spns24 or degraded by S1P phosphatases and/or S1P lyase (S1PL),6 a key enzyme involved in the terminal breakdown of S1P into hexadecenal and ethanolamine phosphate in mammalian tissues.7 ,8 Our preliminary studies showed increased expression of S1PL in lungs from patients with IPF and mice challenged with bleomycin or radiation, which may represent a natural mechanism to partly offset SphK1-mediated S1P production and reduce pulmonary fibrosis.5 However, the role of S1PL and its regulation in pulmonary fibrosis is yet to be defined. In this study we have characterised the effect of partial knockdown of Sgpl1 gene in mice (Sgpl1+/–) on bleomycin-induced lung inflammation and fibrosis, and the role of transforming growth factor (TGF)-β in the upregulation of S1PL in lung fibroblast. Further, S1PL expression in peripheral blood mononuclear cells (PBMCs) from patients with IPF was correlated with pulmonary functions and survival.
Materials and methods
For details regarding materials and methods, see the online supplementary data.
Human subjects
The study population consisted of a cohort of 44 patients evaluated at the University of Chicago. The diagnosis of IPF was established based on American Thoracic Society/European Respiratory Society (ATS/ERS) criteria, which is consistent with recent guidelines. Subjects were followed by physicians according to institutional practices. Pulmonary function tests, chest CT and lung biopsies were performed when clinically indicated. Patients had no evidence of autoimmune syndromes, malignancies, infections or drug or occupational exposures associated with lung fibrosis. All patient information was maintained in a relational database. The study was approved by the institutional review board at each centre and informed consent was obtained from all the patients before blood draw. Demographical and clinical characteristics of the patients in this cohort are provided in online supplementary table S1. Gene annotations were obtained from the Affymetrix NetAffy Analysis Center (http://www.affymetrix.com/). The microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (Accession Number: GSE38958). The frozen lung tissues from controls and human patients with IPF were obtained from the Lung Tissue Research Consortium (funded by NHLBI Lung Tissue Consortium, University of Colorado Health Sciences Center, Denver, Colorado, USA). Real-time PCR was performed to assess expression of target genes using primers as shown in online supplementary table S2.
Results
S1PL expression is increased in lung fibrotic lesions and fibroblasts from patients with IPF and bleomycin-challenged mouse lungs
We have earlier demonstrated enhanced expression of SphK1 in IPF and bleomycin- as well as radiation-challenged mouse lungs,5 and increased S1P level has been implicated in human IPF9 and animal models of pulmonary fibrosis.5 S1P accumulation in pulmonary fibrosis may result from a shift in the balance between its synthesis mediated by SphKs and degradation catalysed by S1P phosphatases and S1PL. To investigate the potential role of S1PL in pulmonary fibrosis, we examined frozen lung tissues from controls and human patients with IPF obtained from the Lung Tissue Research Consortium (Funded by NHLBI Lung Tissue Consortium, University of Colorado Health Sciences Center, Denver, Colorado, USA) as well as bleomycin-induced and radiation-induced murine models of lung fibrosis. Both SGPL1 mRNA and protein expression levels were increased (∼3-fold compared with controls) in IPF lung homogenates (figure 1A, B) and immunostaining of paraffin-embedded lung tissues revealed increased S1PL expression in IPF specimens compared with controls, especially in fibrotic foci (figure 1C). Also, the expression of S1PL in human lung fibroblasts, isolated from patients with IPF, was higher than that from control patients (figure 1D, E).

S1PL is upregulated in lung tissues and fibroblasts obtained from patients with IPF. (A–C) Expression level of S1PL in lung tissues from controls and patients with IPF. (A) Western blot; (B) real-time PCR and (C) immunohistochemistry staining were used to check the expression and location of S1PL in lung tissues from controls and patients with IPF. (D and E) Expression of S1PL in human lung fibroblasts from controls and patients with IPF. (D) Western blot and (E) real-time PCR were used to check the expression of S1PL in human lung fibroblasts from control and patients with IPF. n=5. Data are expressed as mean±SEM of three independent experiments; *p<0.05; p<0.01. α-SMA, α-smooth muscle actin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IPF, idiopathic pulmonary fibrosis; S1PL, sphingosine-1-phosphate lyase.
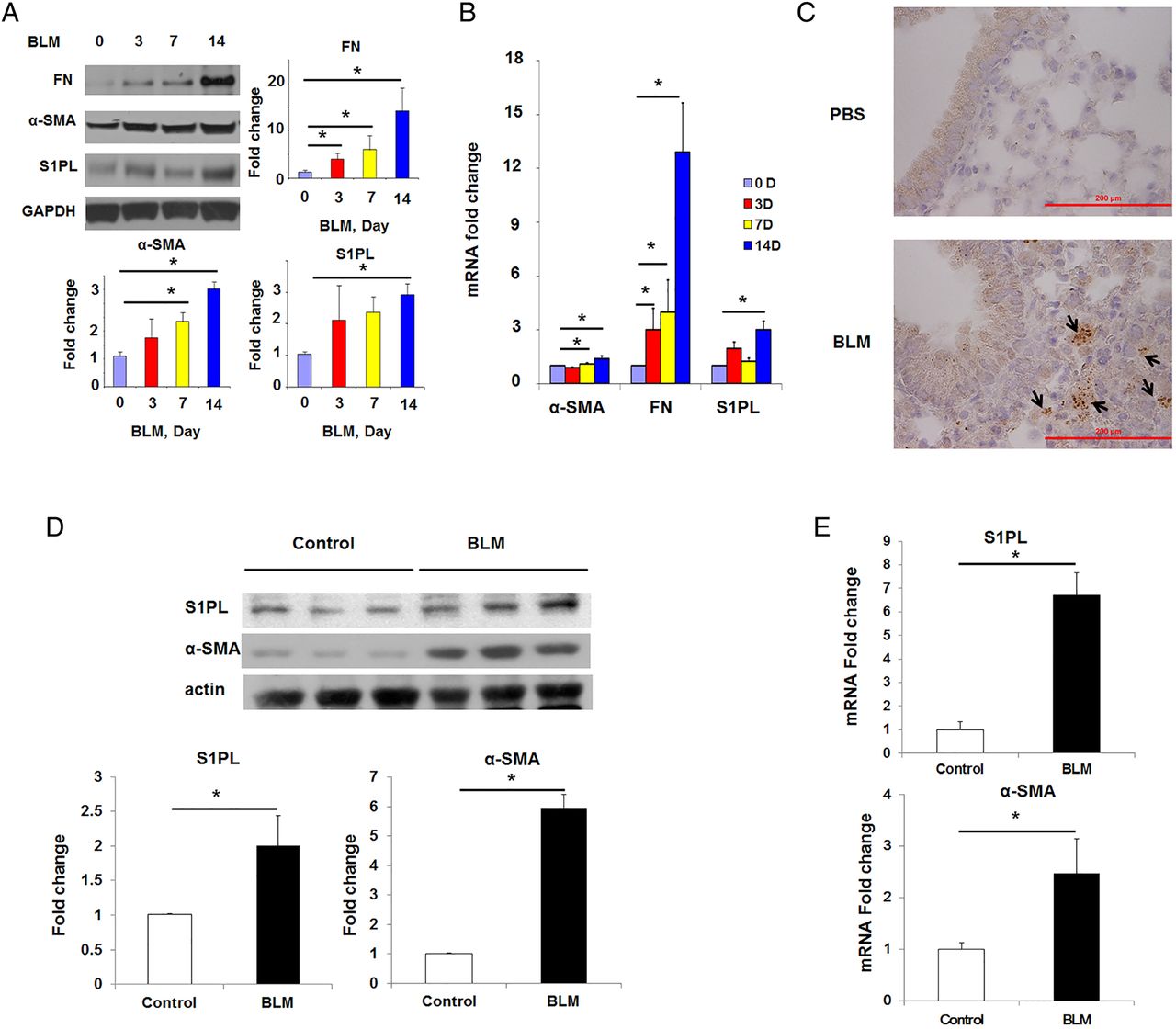
To further validate the enhanced S1PL expression in pulmonary fibrosis, two murine models of pulmonary fibrosis and lung injury were used. Bleomycin challenge increased S1PL protein and mRNA expression in lung tissue homogenates (at day 14) with concomitant upregulation of α-smooth muscle actin (α-SMA) and fibronectin (FN) (figure 2A, B), markers of pulmonary fibrosis.10 ,11 Further, immunostaining of paraffin-embedded lung tissues from bleomycin-challenged mice confirmed induction of S1PL expression in fibrotic foci (figure 2C). Similarly, as shown in figure 2D,E, the protein and mRNA expression levels of S1PL in lung fibroblasts isolated from bleomycin-treated mice were significantly increased compared with that from control mice (∼3-fold change). Similar to the bleomycin model, as shown in online supplementary figure S1, thoracic radiation also enhanced S1PL expression during the later stages of pulmonary fibrosis (14–17 weeks post-irradiation).

S1PL is upregulated in lung tissues and fibroblasts obtained from bleomycin-challenged mice. (A–C) Expression level of S1PL in lung tissues from mice treated with BLM (2 U/kg body weight) for different time (0–14 days). (A) Western blot (0–14 days); (B) real-time PCR (0–14 days) and (C) immunohistochemistry staining (14-day post challenge) were used to check the expression and location of S1PL in lung tissues from mice at different time point post-BLM treatment. n=4 per group. (D and E) Expression of S1PL in mouse lung fibroblasts from mice that have undergone BLM treatment for different time. (D) Western blot and (E) real-time PCR were used to check the expression of S1PL in lung fibroblasts from controls and BLM-treated mice. Control, 0 day post-treatment; BLM, 14 days post-treatment. Data are expressed as mean±SEM of three independent experiments; *p<0.05. α-SMA, α-smooth muscle actin; BLM, bleomycin; FN, fibronectin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; S1PL, sphingosine-1-phosphate lyase.
TGF-β-induced S1PL expression in lung fibroblast via activation of Smad signalling
Since TGF-β is a key cytokine in fibroblast proliferation and differentiation, we evaluated the effect of TGF-β on S1PL expression in human lung fibroblasts. TGF-β challenge dramatically enhanced the protein and mRNA expressions of S1PL in human lung fibroblast (figure 3A, B), and pretreatment with anti-TGF-β antibody abolished TGF-β-mediated expression of S1PL (figure 3C). As TGF-β regulates the expression of number of genes via Smad pathway,1 we investigated the role of Smad2/3 in TGF-β-induced S1PL expression. As shown in figure 4A,B, transfection of human lung fibroblasts with Smad3 small interfering RNA (siRNA) downregulated ∼60% of Smad3 mRNA level, and attenuated TGF-β-induced mRNA and protein levels of S1PL, α-SMA and FN in human lung fibroblasts. In silico analysis revealed presence of at least two consensus sequences for Smad2/3 binding in hSGPL1 promoter region (at −75 bp and −143 bp upstream of transcription start site) (figure 5A). The binding of Smad3 to SGPL1 promoter regions containing Smad consensus sequences was evaluated using chromatin immunoprecipitation (ChIP) assay, and the quality of the ChIP DNA immunoprecipitated by Smad3 antibody was confirmed by the real-time PCR amplification of hSGPL1 promoter region. In Smad immunoprecipitation experiments, the binding of Smad3 to the consensus sites at −75 bp and −143 bp was dramatically increased post-TGF-β challenge (figure 5A); whereas rabbit IgG control failed to pull down the promoter region of hSGPL1 and was not affected by TGF-β challenge (figure 5A). Further, mutation of the two Smad binding sites (−75/−79 and −143/−147) almost completely blocked TGF-β-induced hSGPL1-luciferase reporter activity (figure 5B). These results show that TGF-β-induced S1PL expression is dependent on Smad2/3 binding to Smad consensus sequences on the promoter of hSGPL1.
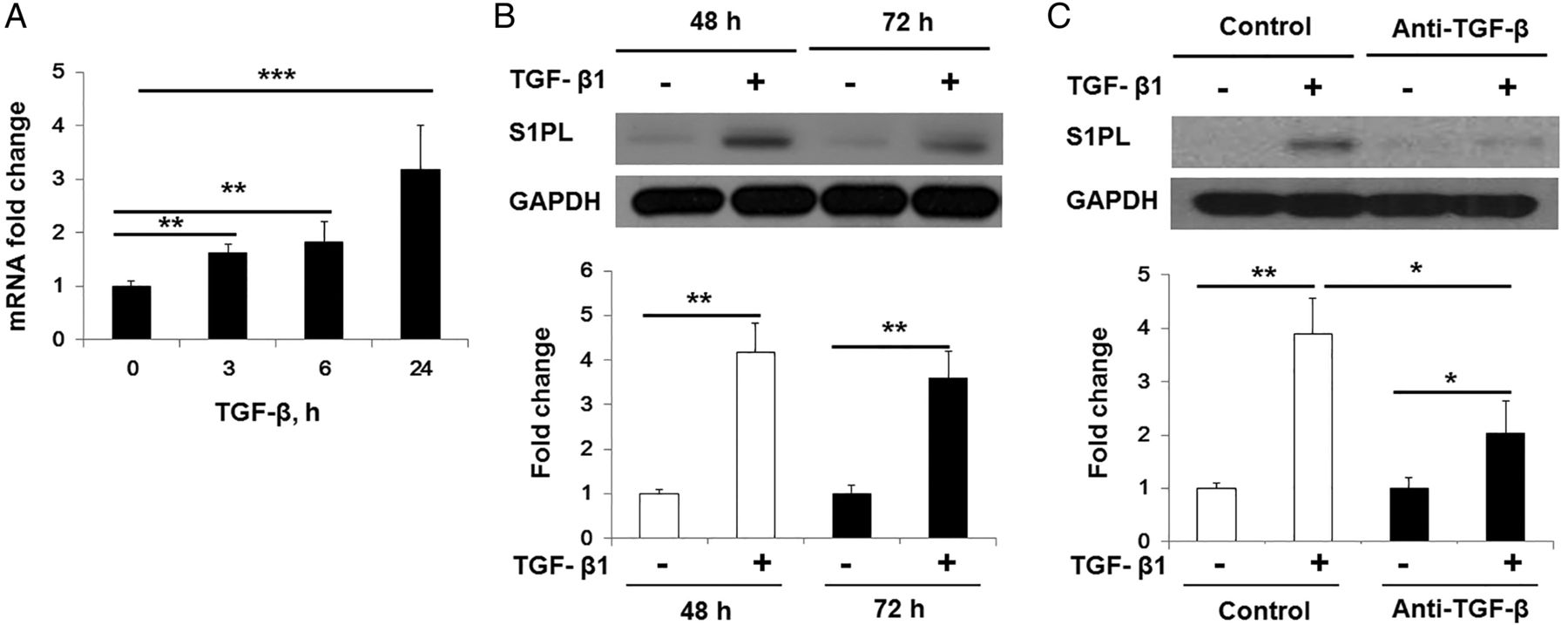
TGF-β-induced S1PL expression in human lung fibroblasts. (A) TGF-β (5 ng/mL) increased the mRNA level of SGPL1 in human lung fibroblasts at different time points (0–24 h). (B) TGF-β (5 ng/mL) increased the protein level of S1PL in human lung fibroblasts at 48 and 72 h. (C) TGF-β (5 ng/mL) induced expression of S1PL (48 h) in human lung fibroblasts was blocked by pretreatment of anti-TGF-β antibody (10 μg/mL). Data are expressed as mean±SEM of three independent experiments; *p<0.05; **p<0.01; ***p<0.001. S1PL, sphingosine-1-phosphate lyase; TGF, transforming growth factor.

Knockdown Smad3-attenuated TGF-β-induced S1PL expression in human lung fibroblasts. (A and B) Smad3 was knocked by treatment of si-Smad3 (100 nM) for 24 h, and cells were followed by treatment of TGF-β (5 ng/mL, 48 h). The mRNA (A) and protein (B) level of Smad2/3, FN, α-SMA and S1PL in human lung fibroblasts were analysed. Data are expressed as mean±SEM of three independent experiments; *p<0.05; **p<0.01. α-SMA, α-smooth muscle actin; FN, fibronectin; S1PL, sphingosine-1-phosphate lyase; TGF, transforming growth factor.
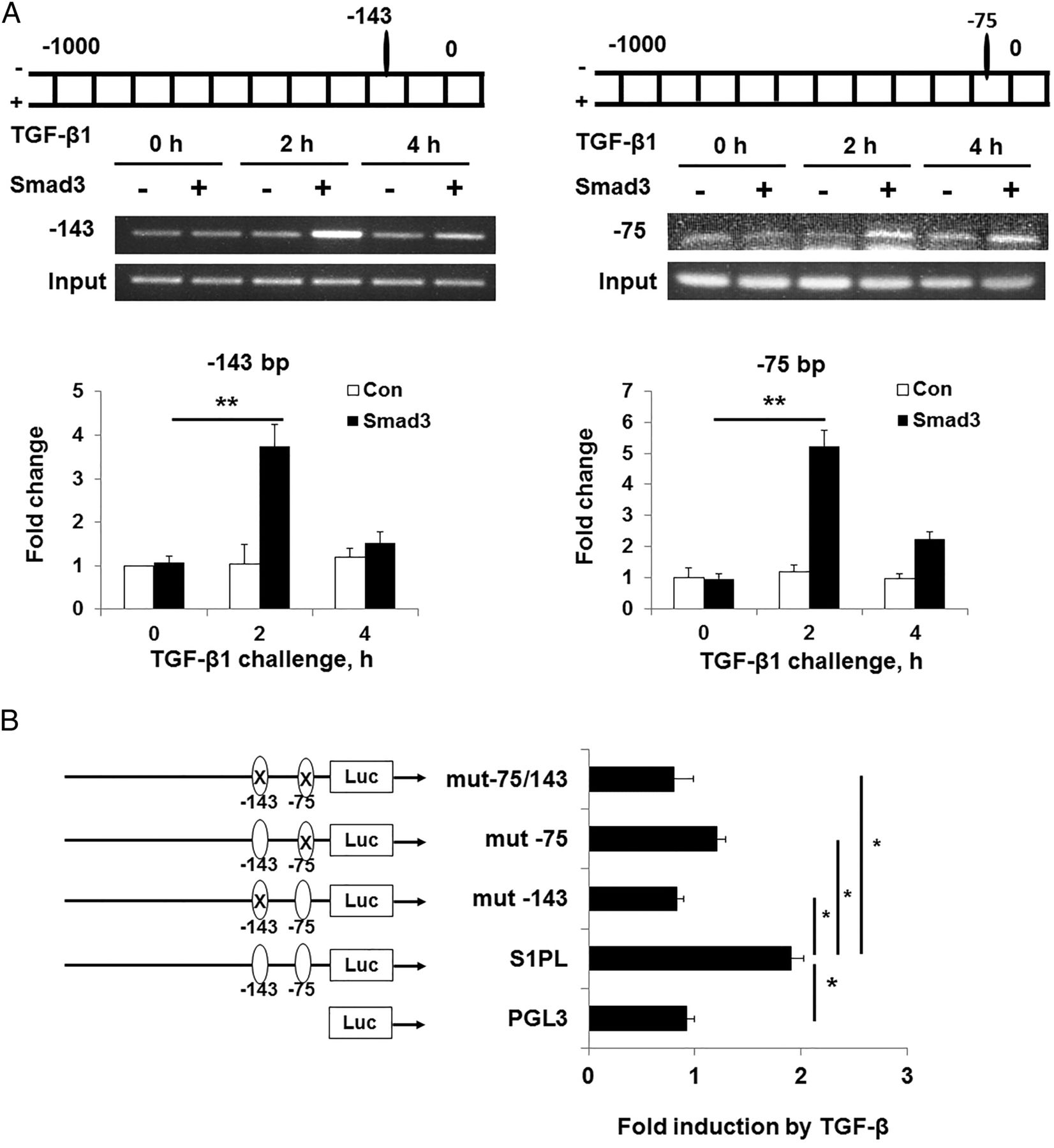
Binding of Smad3 to SGPL1 promoter regions was essential to TGF-β-induced S1PL expression in human lung fibroblasts. (A) TGF-β induced the binding of Smad3 to the SGPL1 promoter. Human lung fibroblasts were treated with TGF-β (5 ng/mL, 0–6 h), and the binding of Smad3 in the SGPL1 promoter sequence were analysed by ChIP assay. (B) Mutation of the Smad3 binding site in the SGPL1 promoter area attenuated TGF-β-induced promoter activity. Mutation of Smad3 binding site in the SGPL1 promoter and the promoter activity were assayed as described in methods section. Data are expressed as mean±SEM of three independent experiments; *p<0.05; **p<0.01. ChIP, chromatin immunoprecipitation; Luc, luciferase activity; PGL3, backbone plasmid of promoter plasmid; S1PL, sphingosine-1-phosphate lyase; TGF, transforming growth factor.
Overexpression of S1PL attenuates TGF-β-induced expression of FN and α-SMA via autophagy in human lung fibroblasts
Next, we investigated the effect of overexpression of S1PL on TGF-β-induced expression of extracellular matrix (ECM) proteins, markers of lung fibroblasts differentiation. Infection of primary human lung fibroblasts with adenoviral construct of hSGPL1 (5 multiplicity of infection (MOI), 48 h) resulted in increased expression of S1PL, and decreased expression of FN and α-SMA in response to TGF-β as compared with control cells infected with adenoviral vector backbone (figure 6A–C). A recent study showed that TGF-β-induced activation and differentiation of lung fibroblasts was regulated by autophagy, as knockdown of the autophagy marker protein LC3 or beclin 1, augmented the TGF-β-induced expression of FN and α-SMA.12–14 Transfection of human lung fibroblasts with adenoviral construct of hSGPL1 enhanced the expression of S1PL as well as LC3 and beclin 1, and reversed TGF-β-induced decrease of LC3 expression and autophagosome formation in human lung fibroblasts (figure 6D–F and see online supplementary figure S2). S1P plays a critical role in TGF-β-induced activation and differentiation of lung fibroblasts;5 therefore, we determined the effect of S1PL overexpression on intracellular level of S1P. As shown in figure 6G, H, overexpression of S1PL abolished TGF-β-induced increase in intracellular S1P level; however, it did not alter the levels of ceramide and sphingosine (data not shown). Similar to TGF-β, the S1P-induced protein and mRNA expression of FN, α-SMA was also suppressed by overexpression of S1PL in human lung fibroblasts (figure 7A and see online supplementary figure S3). Further, S1P challenge also attenuated LC3 expression and autophagosome formation in human lung fibroblasts (figure 7B and see online supplementary figure S4), and overexpression of S1PL blocked S1P-induced attenuation of LC3 mRNA and protein expression (figure 7B and see online supplementary figure S3). Rapamycin and chloroquine have been reported to induce LC3 II accumulation in different cell types through induction of autophagy or inhibition of autophagy flux.15 Similar to S1PL, both rapamycin (see online supplementary figure S5) and chloroquine (see online supplementary figure S6) inhibited TGF-β-induced human lung fibroblast differentiation. Additionally, pretreatment of human lung fibroblasts with chloroquine further increased the effect of S1PL-mediated accumulation of LC3 II expression and inhibition of fibroblast differentiation (see online supplementary figure S7), while knockdown of beclin 1 with small hairpin RNA attenuated S1PL-mediated expression of LC3 II, beclin 1 and restored the inhibition of fibroblast differentiation (see online supplementary figure S8). Further, TGF-β-mediated activation of MAPKs, critical regulators in fibrogenesis, was dramatically inhibited by increased expression of S1PL in human lung fibroblasts (see online supplementary figure S9). Together, these results suggest that overexpression of S1PL attenuates TGF-β-induced intracellular S1P levels, MAPK activation, expression of FN and α-SMA, while it restores autophagy in lung fibroblasts.

Overexpression of S1PL blocked TGF-β-induced differentiation in human lung fibroblasts by decreasing intracellular S1P level and restoring autophagy. (A–C) Overexpression of S1PL blocked TGF-β-induced differentiation in human lung fibroblasts, and the expression of α-SMA and FN was assayed by western blot. Representative western blot (A) and quantification of FN (B) and α-SMA (C). (D–F) Overexpression of S1PL blocked TGF-β effect on autophagy characterised by the expression of LC3 and beclin 1. Representative western blot (D) and quantification of beclin 1 (E) and LC3 II (F). (G and H) Overexpression of S1PL decreased intracellular levels of S1P (G) and DH-S1P (H) in human lung fibroblasts. S1P and DH-S1P level in the human lung fibroblasts with and without overexpression of S1PL were extracted and analysed as described in methods section. Data are expressed as mean±SEM of three independent experiments; *p<0.05. α-SMA, α-smooth muscle actin; DH-S1P, dihydrosphingosine 1-phosphate; FN, fibronectin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; n.s, non-significant; S1PL, sphingosine-1-phosphate lyase; TGF, transforming growth factor.

Overexpression of S1PL blocked S1P-induced differentiation in human lung fibroblasts through restoring autophagy. (A) Overexpression of S1PL blocked S1P-induced differentiation in human lung fibroblasts, and the expression of α-SMA and FN was assayed by western blot. (B) Overexpression of S1PL blocked S1P effect on autophagy characterised by the expression of LC3 II and beclin 1. Data are expressed as mean±SEM of three independent experiments; *p<0.05. α-SMA, α-smooth muscle actin; FN, fibronectin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; n.s, non-significant; S1PL, sphingosine-1-phosphate lyase.
Genetic deletion of S1PL (Sgpl1+/−) decreases autophagy in mouse lung fibroblasts
The role of S1P in autophagy is controversial. Earlier studies indicate that S1P is an inducer16–20 or inhibitor of autophagy.21–23 However, the role of S1P as a regulator of autophagy in pulmonary fibrosis is unclear. Therefore, we investigated the status of autophagy in mouse lung fibroblast isolated from wild-type and S1PL-deficient (Sgpl1+/−) mice. As shown in online supplementary figure S10, the expression of beclin 1 and LC3 and the number of autophagosomes in lung fibroblast isolated from Sgpl1+/− mice were significantly lower than that from wild-type mice. Further, chloroquine, treatment increased the autophagosome numbers in lung fibroblasts from wild-type mice; however, the autophagosomes in lung fibroblast isolated from Sgpl1+/− mice were lower than that from wild-type mice (see online supplementary figure S11). These data suggest that genetic deletion of Sgpl1 attenuates autophagy in mouse lung fibroblasts.
Genetic deletion of S1PL (Sgpl1+/−) accentuates bleomycin-induced pulmonary fibrosis in mice
To further validate the role of S1PL in pulmonary fibrosis, we used Sgpl1+/− (heterozygous) mice as deletion of both alleles of Sgpl1 gene results in severe defects in vascular development and mortality within 4–6 weeks.8 S1PL activity in total lung homogenates from Sgpl1+/− mice was lower than wild-type mice.8 S1P levels, as measured by liquid chromatography-tandem mass spectrometry, revealed a small but significant increase in lung tissues and bronchoalveolar lavage (BAL) fluids, but not in plasma of Sgpl1+/− mice (data not shown). The effect of S1PL deficiency on bleomycin-induced lung injury and fibrosis was evaluated by H&E staining for histopathology and Masson's trichrome staining for collagen in lung sections. Bleomycin challenge significantly increased lung injury and collagen deposition in wild-type mice on day 21, which was further enhanced in bleomycin-challenged Sgpl1+− mice (figure 8A, B). Histopathological quantification for pulmonary fibrosis (Ashcroft score) showed a significant increase of fibrogenesis in Sgpl1+/− mice compared with control group (figure 8C). Importantly, Sgpl1+/− deficiency alone had no significant effect on lung collagen content or histopathological evidence of pulmonary fibrosis in the absence of bleomycin insult. Further, bleomycin-induced lung collagen content, and TGF-β1 levels in BAL fluids were markedly higher in Sgpl1+/− mice compared with wild-type mice (figure 8D, E). Additionally, bleomycin induced significant increase in mRNA levels of several markers of pulmonary fibrosis, including TGF-β, collagen 1A1, FN and α-SMA, on day 21 post-bleomycin challenge in wild-type mice, and the effect of bleomycin was even more pronounced in Sgpl1+/− mice (figure 8F). At the protein level, Sgpl1+/− deficiency significantly increased bleomycin-induced expression of FN and α-SMA, and attenuated the expression of LC3 II and beclin 1 (figure 8G).

Sgpl1+/− deficiency accentuates BLM-induced lung fibrosis. Sgpl1+/− or WT (129SV) mice (male, 8 weeks) received BLM (2 U/kg in 50 µL PBS) or PBS intra-tracheally and were sacrificed on day 21. Lungs were removed, embedded in paraffin and cut into 5 µm sections for staining. (A) Representative H&E photomicrographs of lung sections obtained from Sgpl1+/− and WT mice with/without BLM challenge (black arrows showing injury area). Original magnification, ×10. Scale bar: 200 µm. (B) Representative images of trichrome staining of lung sections obtained from Sgpl1+/− and WT mice with/without BLM challenge (blue arrows showing blue of collagen deposition area). Upper panel, original magnification, ×4; Scale bar: 1 mm. Lower panel, amplification from box in upper panel, original magnification, ×10; Scale bar: 200 µm. (C) Ashcroft score of lung sections, (D) acid-soluble collagen in lung tissue, (E) TGF-β1 level in BAL fluids, (F) mRNA level of TGF-β1, α-SMA, Col1A1 and FN, (G) protein levels of beclin 1, LC3 II, α-SMA and FN from Sgpl1+/− and WT mice with/without BLM challenge. Data are expressed as mean±SEM; *p<0.05, **p<0.01; n=4–6 per group. α-SMA, α-smooth muscle actin; BLM, bleomycin; Col1A1, collagen 1A1; FN, fibronectin; PBS, phosphate buffered saline; S1PL, sphingosine-1-phosphate lyase; TGF, transforming growth factor; WT, wild-type.
Next, the effect of Sgpl1+/− deficiency on bleomycin-induced lung inflammation was assessed. Bleomycin challenge of wild-type mice significantly increased protein levels in BAL fluids, a marker of lung damage and pulmonary leak, relative to the vehicle-treated group; however, the increase in protein levels in BAL fluids of bleomycin-challenged Sgpl1+/− mice were similar to that of wild-type animals at day 14 post-bleomycin challenge (see online supplementary figure S12A). Furthermore, influx of cells into alveolar space in response to bleomycin was marginally increased in Sgpl1+/− mice compared with wild-type mice at day 14 post-bleomycin challenge (see online supplementary figure S12B). Differential cell counts of BAL fluids showed that bleomycin-induced infiltration of macrophages and lymphocytes, but not neutrophils, into alveolar space was higher in Sgpl1+/− mice compared with wild-type mice at day 14 post-bleomycin challenge (see online supplementary figure S12C–E), while interleukin (IL)-6 levels were similar in BAL fluids in both the groups (see online supplementary figure S12F). Together, these results demonstrate that Sgpl1+/− deficiency accentuates bleomycin-induced pulmonary fibrosis.
Correlation between SGPL1 expression in PBMCs, lung function and survival in patients with IPF
To study the role of S1PL in human pulmonary fibrosis, we compared the SGPL1 mRNA expression levels in PBMCs with lung function and survival in patients with IPF. Microarray analysis of PBMCs of patients with IPF (n=44) showed that the mRNA expression levels of SGPL1 positively correlated with diffusing capacity of the lung for carbon monoxide (DLCO) (figure 9A). There is an inverse correlation between DLCO and IPF disease severity24 and our analysis revealed that lower expression of SGPL1 in patients correlated with relatively severe IPF (figure 9A). Next, patients were classified as having a high-level or low-level mRNA expression of SGPL1, with the median of expression as the threshold value. We tested the correlation between SGPL1 expression and survival to classify patients into prognostic groups. Kaplan–Meier survival analysis comparing patient groups demonstrated a marginal increase in survival for patients with high expression levels in SGPL1 mRNA (figure 9B). Together, these data suggested that SGPL1 mRNA expression in PBMCs may serve as potential indicator of survivability in IPF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of SGPL1 correlates with the severity of disease and survival of patients with idiopathic pulmonary fibrosis (IPF). (A) Expression level as a function of DLCO for SGPL1 in IPF. Y-axis denotes the relative mRNA expression level. Each point represents the value from one patient with IPF. Y-axis denotes the relative mRNA expression level. The p value for each gene is measured by Spearman’s rank correlation test. Correlation coefficient: ρ=0.316. (B) Kaplan–Meier curves for patients with IPF high and low gene expression level of SGPL1. Patients were classified as having a high-level or a low-level mRNA expression, with the median of the expression as the threshold value. The p values are measured by log-rank test.
Discussion
Idiopathic lung fibrosis is a chronic fatal lung disease of unknown aetiology with limited effective therapies. The pathogenesis of IPF is characterised by fibroblast to myofibroblast differentiation, epithelial to mesenchymal transition, deposition of ECM proteins such as FN, enhanced expression of α-SMA and excess deposition of collagen in lungs.1 Accumulation of myofibroblasts results in gradual but excessive scarring of the lung tissues that undermines essential lung functions leading to eventual respiratory failure. Therefore, identification of new targets against fibroblast differentiation and accumulation during pulmonary fibrosis is critical to a better understanding of the pathobiology of pulmonary fibrosis and development of new strategies for treatment. Here, we report that S1PL, one of the key enzymes involved in terminal degradation of S1P to hexadecenal and ethanolamine phosphate in mammalian cells, has a regulatory role in the development of pulmonary fibrosis in both human IPF and bleomycin-induced murine model of fibrosis.
A role for S1P and S1P signalling in pulmonary fibrosis is emerging in human IPF and animal studies. S1P is a bioactive sphingolipid, which is generated by phosphorylation of sphingosine, catalysed by SphK1/2 in mammalian cells.4 S1P levels in most of the mammalian cells are lower compared with plasma, platelets and erythrocytes.4 In most of the cells, intracellularly generated S1P is either metabolised to sphingosine by S1P phosphatases or to ethanolamine phosphate and hexadecenal by S1PL.8 Additionally, S1P is also transported to outside of cells by S1P transporters such as ATP-binding cassettes and Spns2.25 A recent study has demonstrated the increased expression of SphK1 mRNA and S1P levels in lung tissues from patients with IPF, and downregulation of SphK1 as well as S1PR2 and S1PR3, but not S1PR1, with siRNA-attenuated TGF-β-mediated upregulation of FN and α-SMA in human lung fibroblasts, suggesting involvement of S1P signalling in regulating TGF-β-induced fibroblast to myofibroblast differentiation.5 Our recent work, using the mouse model of radiation-induced and bleomycin-induced pulmonary fibrosis, demonstrated involvement of SphK1/S1P signalling axis in the development of pulmonary fibrosis.5 ,9 In the radiation model, inhibition of serine palmitoyltransferase delayed the onset of pulmonary fibrosis through the negative regulation of SphK1 expression.26 Interestingly, genetic deletion of Sphk1 or inhibition of SphK1 activity with a small molecular weight inhibitor-attenuated S1P generation, TGF-β secretion and pulmonary fibrogenesis in the murine model of bleomycin-induced lung fibrosis.5 In vitro, bleomycin-mediated enhanced expression of SphK1 was TGF-β-dependent, as addition of anti-TGF-β antibody attenuated the increases in SphK1, α-SMA and FN expression in human lung fibroblasts.5 Here, we observed that both bleomycin and TGF-β increased the Sgpl1 mRNA and protein expression via transcriptional regulation of Smad2/3 in human lung fibroblast. Our present findings combined with our earlier observation on TGF-β-induced expression of SphK1 strongly suggest that TGF-β-mediated regulation of S1P levels is dependent on the balance between SphK1 and S1PL expression and activities in human lung fibroblast. Interestingly, TGF-β regulates S1P levels in human fibroblast,5 and its expression is modulated by S1P, which is a balance between SphK1 and S1PL expression and activity. Our current results show that overexpression of SGPL1 in human lung fibroblasts blocks TGF-β-mediated increase in S1P, expression of FN and α-SMA and fibroblast differentiation. However, the increased expression of S1PL in lung tissues from patients with IPF and bleomycin-treated mice may contradict the role of S1P in pulmonary fibrosis. In the model of radiation-induced pulmonary fibrosis, increased S1PL expression was not accompanied by enhanced S1PL enzyme activity.26 It is unclear if S1PL undergoes any post-translation modification such as nitration, phosphorylation or ubiquitination to bleomycin or radiation challenge, which could modulate its ability to degrade S1P.
An interesting and important finding of current study is that S1PL/S1P signalling axis mediates regulation of autophagy in human lung fibroblasts. In the current study, we provide evidence for decreased expression of LC3 II, beclin 1 and autophagosome formation in isolated fibroblasts from Sgpl1+/− mouse lungs compared with the wild-type, and exposure of normal human lung fibroblasts to TGF-β or S1P attenuated LC3 II expression and autophagosome formation. Further, overexpression of hSGPL1 significantly increased beclin 1 and LC3 II levels in the absence or presence of TGF-β and S1P. These results for the first time demonstrate the influence of S1P and its role in regulating LC3 II and beclin 1, markers of autophagy, in lung fibroblast. There is increasing evidence for insufficient autophagy in IPF. While lung tissues from patients with IPF showed decreased autophagic activity as assessed by LC3, p62 expression and the number of autophagosomes,12 and based on the in vitro results from human fibroblasts isolated from patients with IPF and administration of rapamycin,27 an inducer of autophagy to bleomycin-challenged mice, it was concluded that autophagy is not activated in the setting of IPF,13 and insufficient autophagy was involved in fibroblast to myofibroblast differentiation in human IPF.12 These results indicate that impairment of autophagy in IPF lung might be TGF-β-dependent, and the TGF-β effect on autophagy could be related in part to S1P production, as S1P attenuates LC3 expression in human lung fibroblasts (figure 7). The present results suggest that increased intracellular S1P level is essential for the fibroblast differentiation and autophagy, and suppression of intracellular S1P level through overexpression of S1PL enhanced autophagy and inhibited fibroblast differentiation. Further, our current studies with chloroquine and rapamycin (see online supplementary figure S5–S7) indicate that the effect of S1PL on autophagy in lung fibroblast may be through the regulation of LC3 II and beclin 1 expression rather than autophagy flux. Further studies are necessary to delineate the mechanism(s) involved in S1P-mediated modulation of autophagy in fibroblasts.
In summary, we have identified that S1PL is a novel endogenous suppressor of human lung fibroblast differentiation and development of pulmonary fibrosis in bleomycin-challenged mouse. We have shown increased expression of S1PL in lung tissues and fibroblasts from patients with IPF and bleomycin-challenged or radiation-challenged mice, and identified that S1PL inhibited lung fibrosis through regulating intracellular S1P level and autophagy in lung fibroblast. In vitro TGF-β-stimulated S1PL expression was transcriptionally regulated by Smad2/3 signalling in human lung fibroblast. This study identifies for the first time, using Sgpl1+/− mice, that knockdown of the S1PL accentuated bleomycin-induced pulmonary fibrosis in mice, and the clinical investigation also indicated that the expression of S1PL in PBMCs directly correlated to the severity of fibrosis in human. As differentiation of lung fibroblast to myofibroblast is critical to the development of pulmonary fibrosis, the role of S1PL and S1P signalling in fibrogenesis needs to be further investigated to better understand the pathobiology of IPF.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Contributors VN and JGNG supervised the overall project and experiments; VN, LSH and BM, designed experiments; LSH, BM, WM and AH performed animal experiments; LSH, JTT, LX, JC, GW and CF-B performed in vitro experiments; EVB and IG performed liquid chromatography-tandem mass spectrometry; LSH, EVB and IG analysed the mass spec data and sphingosine-1-phosphate levels; TZ, WZ, S-FM, IN, SPR and JGNG collected and analysed the human patient samples and LSH and VN wrote the manuscript.
Funding This work was supported by NIH/NHLBI grant P01 HL 98050 to VN.
Competing interests None declared.
Patient consent Obtained.
Ethics approval University of Chicago.
Provenance and peer review Not commissioned; externally peer-reviewed.