Article Text

Abstract
Macrophages are the most numerous immune-cells present in the lung environment under homoeostatic conditions and are ideally positioned to dictate the innate defence of the airways. Pulmonary macrophage populations are heterogeneous and demonstrate remarkable plasticity, owing to variations in origin, tissue residency and environmental influences. Lung macrophage diversity facilitates considerable specialisation, aids efficient responses to environmental signals and allows rapid alterations in phenotype and physiology in response to a plethora of cytokines and microbial signals. This review describes pulmonary macrophage origins, phenotypes, roles in diseases of the airways and implications for the treatment of respiratory disease.
- Innate Immunity
- Macrophage Biology
- Idiopathic pulmonary fibrosis
- Cystic Fibrosis
- COPD Exacerbations
- Bacterial Infection
- Asthma Mechanisms
- Respiratory Infection
Statistics from Altmetric.com
- Innate Immunity
- Macrophage Biology
- Idiopathic pulmonary fibrosis
- Cystic Fibrosis
- COPD Exacerbations
- Bacterial Infection
- Asthma Mechanisms
- Respiratory Infection
Introduction
Metchnikoff first described macrophages in 1892 on the basis of their ability to phagocytose microorganisms. Since then our understanding of this key player of the immune system has expanded tremendously.1 Macrophages are a critical component of the primary innate immune response and are present in almost all tissues of the body.2 In the lung, they are the most abundant immune-cell type present under homoeostatic conditions. Pulmonary macrophages form a heterogeneous population of immune cells that fulfil a variety of specialised functions, including maintenance of pulmonary homoeostasis, removal of cellular debris, immune surveillance, microbial clearance, responses to infection and the resolution of inflammation. This remarkable plasticity originates from variations in origin, tissue residency and environmental influences. Macrophages exhibit significant functional and phenotypical specialisation, allowing efficient responses to environmental signals and enabling rapid alterations in phenotype and physiology. This review describes pulmonary macrophage phenotypes and functions. Particular emphasis is given to pulmonary diseases where macrophages may play a central role and therefore present a feasible target for therapy development.
Pulmonary macrophage phenotypes and origins
At least two distinct macrophage populations exist in the lung at homoeostasis, termed alveolar macrophages (AMs) and interstitial macrophages (IMs), respectively. Each are characterised by their unique location, properties and functions.3 AM and IM populations may be identified on the basis of their expression patterns of the integrins CD11b and CD11c.3 AMs, which are located in the airway lumen are characterised by high expression of CD11c but lack CD11b expression, distinguishing them from macrophages present in other tissue compartments. Conversely, IMs reside in the lung parenchyma, express high levels of CD11b and low levels of CD11c. AMs play a central role in maintaining immunological homoeostasis and host defence in the lung and are inherently suppressive, whereas IMs are thought to have a regulatory function within the lung tissue. While AMs exert their regulatory effects via non-specific lines of defence (such as high phagocytic ability, the secretion of antimicrobials, nitric oxide (NO), tumour necrosis factor (TNF)-α and interferon (IFN)-γ), it has been suggested that IMs have a greater propensity to release specific cytokines associated with the adaptive immune response, such as interleukin (IL) 10.4 ,5
Classification of macrophages, according to their activation status has broadly been defined as M1 and M2, analogous to the T helper cells Th1/Th2 paradigm. M1 macrophages (also termed classically activated macrophages) respond to IFN-γ, lipopolysaccharide and/or TNFα, produce proinflammatory cytokines, direct destruction of intracellular pathogens and promote a local Th1 environment. M2 macrophages (also known as alternatively activated macrophages), represent a more diverse phenotype and are characterised by their participation in type 2 immune responses. M2 macrophages can be divided further into M2a, M2b and M2c subsets. These subsets facilitate parasite encapsulation and destruction (M2a), immunoregulation (M2b) and tissue remodelling and matrix deposition (M2c). M2 macrophages have also been associated with tumour progression.6 Similar to Th1/Th2 lymphocyte polarisation, the switch to an M1 or an M2 phenotype has been associated with specific transcriptional control.7 The transcription factor IFN regulatory factor 5 has been shown to control the M1 phenotype in human and murine studies, and dictate proinflammatory cytokine production in these cells.8 Investigations into the respective roles that M1 and M2 macrophages may play in respiratory diseases has proved complex. Macrophages are highly plastic, and during pulmonary inflammatory disease, are capable of finely tuning their activity and developing mixed phenotypes in response to the local microenvironment. It is apparent that during disease, rather than existing at the extreme points of the M1/M2 spectrum, macrophage phenotype is tailored to meet the immediate immunological need. Importantly, data defining the M1/M2 paradigm are largely based on in vitro studies using monocytes isolated from peripheral blood and care should be taken in extrapolating phenotypical activity from these cell culture studies to activation status during disease progression and within airways of patients. Relating to this, a new consensus is building in terms of macrophage nomenclature, with standards proposed which define macrophages based on defined criteria, such as macrophage source, definition of the activators, and an agreed collection of surface/intracellular markers.9 ,10
More recently, the importance of macrophage function in disease has focused on developmental origins. The prevailing dogma in macrophage biology has been that of the mononuclear phagocyte system. In this model, the majority of tissue macrophages are derived from monocyte precursors present in the circulation and the bone marrow.1 ,11 However, recent mechanistic work in murine models has clarified our understanding of macrophage ontogeny and has challenged the importance of the mononuclear phagocyte system paradigm in pulmonary immunity. It is becoming clear that rather than being recruited from macrophage precursors in the blood, several tissue-resident macrophage populations, including lung AMs, are maintained in situ.12 ,13 Work using mice that lack haematopoietic stem cells have determined that during embryonic development, yolk sac macrophages give rise to macrophages in the lung independently of haematopoietic cells and are established prenatally.14 Similarly, Hashimoto et al showed that monocytes do not make a significant contribution to tissue macrophage populations under homoeostatic conditions and furthermore, following depletion of lung macrophages, repopulation of the lung occurred by proliferation in situ, rather than replacement from the bone marrow.15 These data represent a seismic shift in our understanding of the importance of macrophage phenotypes, origins and lung innate immunity and have the potential to inform future therapeutic strategies directed at macrophage-driven processes. Furthermore, these observations highlight the need to study specific macrophage populations in context, as differences in tissue specificity and developmental origins make direct comparisons of different macrophage populations problematic. The multiple layers of phenotypical diversity (figure 1), combined with the strategic positioning at the interface of the airways and the environment, results in complex contributions of macrophages to diseases of the airways such as pulmonary infection, COPD, cystic fibrosis (CF), asthma, pulmonary fibrosis and age related lung disorders (figure 2).
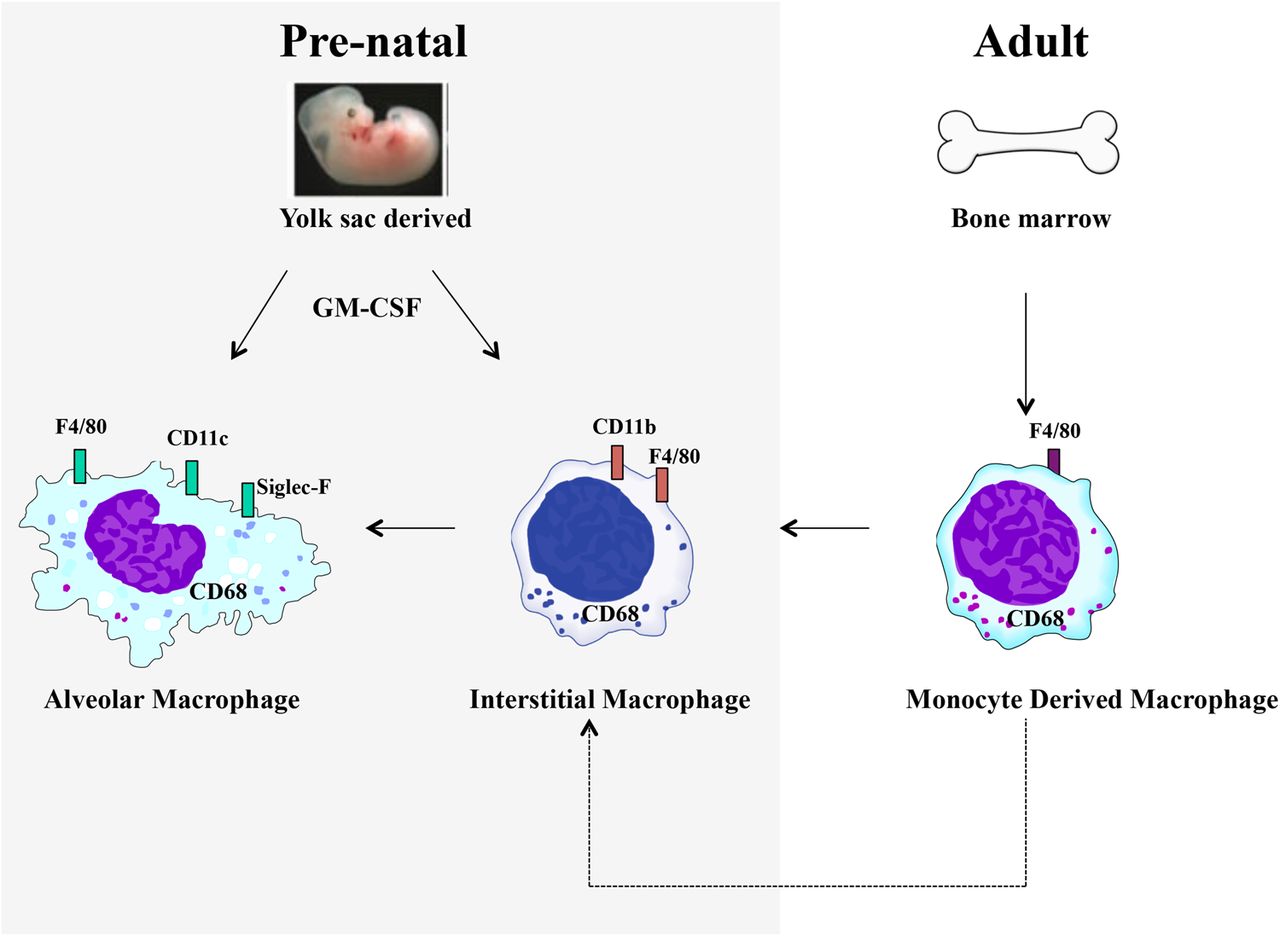
Pulmonary macrophage ontogeny. At least two distinct macrophage populations exist in the lung at homoeostasis, termed alveolar macrophages (AMs) and interstitial macrophages (IMs). AM and IM populations may be identified on the basis of their expression patterns of the integrins CD11b and CD11c. Recent evidence suggests that AMs are long-lived cells that are established prebirth and maintained in situ. Under inflammatory conditions, monocyte-derived macrophages may be recruited from the circulation.

{kind=link}
{kind=link}
Pulmonary macrophages play a central role in lung disease. The plastic nature of pulmonary macrophages, combined with their strategic positioning at the interface of the airways and the environment, results in complex contributions to diseases of the airways.
Macrophage mediation of airway disease
Respiratory tract infections
Respiratory tract infections (RTIs) cover a broad range of causative agents and are a significant complication of chronic pulmonary inflammatory conditions. Interactions between pathogen associated molecular patterns expressed by microbes and pattern recognition receptors expressed by macrophages, can potentially elicit a variety of responses to respiratory infection. Macrophages appear to be central in the pathogenesis of several respiratory tract infections including Mycobacterium tuberculosis, Streptococcous pneumoniae, rhinovirus, influenza and respiratory syncytial virus (RSV).16–18
AMs drive the pulmonary immune response to M. tuberculosis infection. At the early stages, infection promotes an M1 phenotype with expression of high levels of inducible nitric oxide synthase (iNOS) and proinflammatory cytokines.19 Mycobacteria enter the macrophage via interactions with complement receptors, mannose receptors and Fc receptors and replicate slowly and can survive within the cell for many years.19 Once phagocytosed, bacilli survive and either disseminate or remain latent by virtue of their ability to prevent phagolysosome maturation and resist reactive oxygen and nitrogen species. Infected macrophages promote recruitment of Th1 lymphocytes resulting in the formation of a macrophage-rich granuloma. This tissue response is designed to control infection, however, in immunocompromised individuals this response is ineffective and unrestrained, leading to persistent infection and collateral tissue damage. M. tuberculosis infection of human macrophages induces the expression of NO-2 and NO-3; this NO production can also enhance survival of the pathogen.20 Macrophage inhibitory factor has been shown to be a critical component of the innate response controlling disease and individuals with allelic low expression of macrophage inhibitory factor are more susceptible to infection.21 Macrophages play a complex role in the disease and further mechanistic work is required to determine how these cells may be manipulated in order to control infection and disease progression.
S. pneumoniae is a common cause of community-acquired pneumonia and AMs have been shown to play a significant role in clearing the opsonised pathogen.17 Infection is associated with the induction of Th1 cytokines, however the associated macrophage phenotype is unclear. However, transcriptome analysis of Streptococcus pyogenes, a bacterium of the same genus, has been shown to induce a mixed M1 and M2 macrophage phenotype.22 Taut et al demonstrated in a murine model of infection, that populations of resident alveolar and lung parenchymal macrophages are largely replaced by infiltrating peripheral macrophages in response to infection with S. pneumoniae, rather than expansion of resident cells.23 Furthermore, removal of dead/dying cells by AMs results in subsequent impaired phagocytosis and bacterial killing and impaired clearance of S. pneumoniae.24 Moreover, it has been shown that administration of glucocorticoids during active murine pneumonia infection further hinders the ability of AMs to kill bacteria via inhibition of cytokine production.25 This highlights the complexity of glucocorticoid treatment in patients with chronic lung disease and a role for AMs in susceptibility to secondary infection and exacerbation.
Rhinovirus infections are the predominant cause of the ‘common cold’. Infection in susceptible individuals, such as sufferers of asthma, CF and COPD, can result in severe disease exacerbations. Infection with rhinovirus (RV) is sufficient to activate the nuclear factor κ-B pathway in human monocyte derived macrophages and promote TNFα release.18 This implies that macrophages may be a source of proinflammatory cytokines during disease exacerbation in patients with asthma or COPD. Although RV primarily infects the upper airways, distal to the alveoli space, it has been shown experimentally that RV also infects AMs, altering their phenotype. Prior infection with RV reduces the ability of AMs to secrete TNFα and IL-8 in response to toll-like receptor 4 (TLR4) and TLR2 ligands, thus impairing their ability to respond to a subsequent bacterial infection.26 Similarly, RSV has also been associated with asthma exacerbations and morbidity.27 AMs are necessary to illicit the early response to RSV;28 additionally, it has been shown that following RSV infection, AMs are the predominate source of type-1 IFNs that recruit monocytes necessary to mediate antiviral defence in the lungs.29 RSV induces IL-4 and IL-13 production in macrophages and this M2 macrophage phenotype is necessary to control inflammation and promote resolution.30 A key feature of influenza infection is an increased number of AMs in the airway. Work in murine macrophages has demonstrated that influenza infection can result in compromised pathogen clearance, acute inflammation and cell recruitment.31 Infection with the severe acute respiratory syndrome (SARS) coronavirus, the causal agent of SARS, results in a significant accumulation of enlarged M2 macrophages in the lung.32 Spike (S) protein, a major structural component of SARS, induces the conversion of B cells into cells that resemble macrophages, when displayed on recombinant baculovirus.33 Dosch et al34 showed that secreted S protein could activate blood monocytes via TLR2. Furthermore, anti-S antibodies increased infection rates of SARS coronavirus in human monocyte-derived macrophages,35 demonstrating the potential of macrophage-focused therapy during SARS infection
Chronic obstructive lung disease
Compelling evidence supports the hypothesis that innate immune responses are dysfunctional in patients with COPD. Increased numbers of macrophages and macrophage chemoattractants have been found in airways and lung parenchyma of patients suffering from COPD,36 as have enhanced levels of macrophage derived products such as IL-8, TNFα and reactive oxygen species.37 Furthermore, the activity of histone deacetylase, a key enzyme involved in proinflammatory response of macrophages, is impaired in patients with COPD.38 AMs from patients with COPD exhibit impaired induction of TLR2 expression after incubation in vitro with Moraxella catarrhalis, Streptococcus pneumoniae or Haemophilus influenza.39 Furthermore, COPD-AMs exhibit a reduced ability to phagocytose apoptotic epithelial cells,40 eosinophils41 and bacteria.42 Lung parenchymal destruction is a key feature of COPD and macrophages have been shown to directly contribute to this process. In response to IFN-inducible protein 10 and monokine induced by IFN-γ, lung macrophages secrete matrix metalloproteinase (MMP)-12, which is capable of promoting elastin-degradation and tissue destruction.43 AMs derived from COPD-smokers display characteristics of M1 and M2 phenotypes,44 ,45 perhaps indicating that COPD-AMs, although activated in the disease, do not universally conform to the M1/M2 paradigm. Concurrent development of M1 and M2 macrophages is also evidenced by increased levels of iNOS and arginase. Maintenance of AM populations in COPD with skewed expression of M2-related wound healing genes fail to resolve inflammation contributing to detrimental remodelling in the lung. Murine data has indicated that in addition to resident AMs, mononuclear phagocytes, located in the interstitial space, contribute TNFα and IL-6 during cigarette smoke induced emphysema.46
Cystic fibrosis
CF, caused by mutations of the CF transmembrane conductance regulator (CFTR) gene, is characterised by a failure to clear infections with Gram-negative bacteria such as Pseudomonas aeruginosa, Staphylococcus aureus, Burkholderia cepacia and H. influenza. Levels of macrophage-derived products, such as TNFα, IL-1β and IL-8, are elevated in the lungs of patients with chronic CF compared with healthy controls and the switch from IL-10 to inflammatory cytokine production in macrophages and associated lack of suppressor activity is key in the development of CF lung disease.47 Kopp et al48 found that peripheral blood monocyte derived macrophages from patients with CF produced elevated IL-1β, IL-10, monocyte chemoattractant protein (MCP)-1 and IFN-γ after infection with Burkholderia cenocepacia, in comparison to non-CF controls. In contrast, Assani et al49 found that B. cenocepacia infected CF-monocyte derived macrophages produced less IFN-γ; this disparity was attributed to strain-specific differences in B. cenocepacia and/or alterations in patient baseline IFN-γ levels. Peripheral blood monocytes, as opposed to bronchial epithelial cells, are the predominant source of IL-1β in patients with CF and furthermore, specific mutations in the CFTR gene directly affect IL-1β production in murine macrophages upon B. cenocepacia infection.50 ,51 In addition to these alterations in macrophage-derived mediators during the course of CF, changes in macrophage function have also been described. Macrophages from patients with CF and murine macrophages lacking CFTR demonstrate impaired bactericidal killing which may lead to continual stimulation of macrophages contributing to persistent infection and chronic inflammation.52 ,53 Macrophages from patients with CF have also been described to have an inability to recognise pathogens and demonstrated an impaired ability to phagocytose Escherichia coli and P. aeruginosa. Macrophage phenotype appears to be altered during CF pathogenesis as low expression of the alternative activation marker, CD206, has been described in patients suffering from the disease. However, Murphy et al found an increase in the expression of CD206 and arginase in P. aeruginosa-infected patients with CF.54 Furthermore, patients with CF have been shown to have reduced expression of major histocompatibility complex class II (MHCII) molecules in monocytes and macrophages.55 These data indicate that macrophage function/cytokine secretion is significantly impacted during disease progression, raising the intriguing possibility of the development of macrophage directed therapies to treat CF.
Asthma and allergic airways disease
Trials with biologics to target Th2 cytokines in asthma have revealed subpopulations of responders, however the overall outcome was of small clinical impact and therapies have not advanced beyond inhaled steroids and bronchodilators.56 Animal models of allergic airways disease have provided evidence that innate immunity may be of particular importance for allergic diseases of the airways. Indeed, M1 and M2 macrophages are implicated in the pathogenesis of asthmatic disease, highlighting the complexity of the M1/M2 paradigm. High levels of IL-23 and IL-1β produced by M1 macrophages have been shown to prime substantial Th1 and Th17 responses that promote airway neutrophilia and acute airway hyper-responsiveness.57 M2 macrophages respond to IL-14 and IL-13, key cytokines in asthma pathology and furthermore, promote a Th2 environment and airway remodelling.58 Murine studies have demonstrated that CD11b+ myeloid cells in the lung direct the adaptive immune response in allergic airways disease, in part, through the production of signal transducer and activator of transcription-6-inducible chemokines, such as eotaxin, that are essential for the recruitment of Th2 lymphocytes into the airway.59 The removal of AMs in mice worsens lung function and Th2 inflammation; furthermore, transfer of healthy AMs protected against asthma-like symptoms.60 Depletion of AMs during allergic disease delayed inflammatory resolution and there was a decrease in the production of the immune regulatory cytokine IL-27.61 These data suggest that macrophages are capable of dictating the pulmonary immune response to inhaled allergen and furthermore, that allergen exposure may impair the ability of macrophages to maintain lung immune homoeostasis.62 AMs and IMs, have been shown to produce the classic anti-inflammatory cytokine IL-10, reducing responses to inhaled allergen.4 Hypoxia inducible factor-1-alpha (HIF-1α) deficient IMs express reduced IL-10, promoting antigen presentation and dendritic cell proliferation.63 The pharmacological inhibition of HIF-1α was shown to decrease allergic inflammation and eosinophilia via reduced macrophage activity.64 These data indicate a key role for macrophages in the initial response to allergen but also for sustaining disease.
Fungal infection is a concern for immunocompromised individuals. Aspergillus fumigatus is a ubiquitous fungus that is efficiently cleared by immune-competent hosts; however in susceptible individuals exposure can cause invasive pulmonary disease. Experimentally, skewing of AMs to an M2 phenotype has been shown to be protective against A. fumigatus infection raising an attractive therapeutic strategy.65 Furthermore, AMs appear to be critical in regulating the pulmonary innate immune response to A. fumigatus infection, as removal of AMs in mice results in an increased fungal burden.66 Defining the features of AMs that are important to fungal defence will be critical in treating immunocompromised individuals susceptible to infection.
Fibrotic lung disease
Interstitial lung disease is a heterogeneous group of lung disorders characterised by deposition of excess collagenous extracellular matrix in the lungs and the destruction of lung architecture leading to compromised gas exchange. Idiopathic pulmonary fibrosis is the most common form of interstitial lung disease and is associated with a significant decline in quality of life and poor survival rates. Recent evidence suggests that fibrosis in idiopathic pulmonary fibrosis is likely due to a fibroproliferative and dysregulated wound healing cascade that may be driven by macrophages.67 By controlling MMPs, macrophages may also control extracellular matrix turnover.68 Osterholzer et al found that CD11b+, non-resident macrophages and Ly-6Chi monocytes are critical determinants of pulmonary fibrosis induced by selective type II alveolar epithelial cell injury.69 Circulating monocytes from patients with systemic sclerosis exhibit a profibrotic, M2 phenotype.70 Monomeric collagen type I, a collagen breakdown product acting via the scavenger receptor CD204 induces phosphorylation of the protein kinase Akt which shifts AMs to the profibrotic M2 phenotype.71
Several macrophage products have been explored as potential targets to treat pulmonary fibrotic disease. TNFα mRNA has been found to be higher in patients with fibrosis compared with normal controls and furthermore, soluble TNFα receptor effectively prevents silica or bleomycin induced fibrosis when administered therapeutically or prophylactically in mice.72 A randomised controlled trial indicated that some improvement in disease progression could be achieved with anti-TNFα therapy,73 however a subsequent study found that the beneficial effects of anti-TNFα treatment were associated with significant side effects.74
Overexpression of pulmonary IL-1β in rat lungs leads to fibrotic disease75 ,76 and IL-1β expression levels in lung are correlated with the development of pulmonary fibrosis in bleomycin and radiation models of fibrosis. Consistent with these findings, blockade of IL-1β in mice has been shown to ameliorate fibrotic disease.77 Li et al78 showed that epithelial cells and AMs release IL-1 family member, IL-33 in response to bleomycin challenge, leading to M2 macrophage polarisation and production of macrophage and type-2 innate lymphoid cell derived transforming growth factor β1 and IL-13, respectively. Macrophage phagocytosis of asbestos and silica particles results in nicotinamide adenine dinucleotide phosphate oxidase dependent production of reactive oxidation species and IL-1β production and inflammasome activation is sufficient to drive fibrotic disease.77 ,79 Roles for M1 and M2 macrophage subtypes have been described during fibrotic lung processes. M1 cells have been shown to produce extra-cellular matrix (ECM) degrading MMPs, and proinflammatory cytokines during disease73 ,80 ,81 Multiple studies link M2 macrophages to fibrotic processes through the production of chemokines, MMPs, tissue inhibitor of metalloproteinases (TIMPs) and fibronectin.65 ,82–84 However, macrophages with mixed M1/M2 phenotypes have been described during the disease and both subtypes are likely to play a role.
The environment and aging
The primary role of AMs is to maintain the airways by elimination of foreign particulates. AMs exposed to foreign matter phagocytose these particles in vivo and in vitro even in the absence of opsonisation.85 Macrophages may also be activated by ambient particulate matter containing TLR activating microbial products such as lipopolysaccharide, β-glucan, bacteria and fungal spores.86 ,87 Bauer et al88 showed that co-culture of human primary AMs with epithelial cells modifies macrophage responses to O3, enhancing M2 activation status and reducing phagocytic activity. Goto et al89 demonstrated that cytokine production by AM in response to particulate air pollution accelerates monocyte release from the bone marrow. Interestingly, Shaw et al90 showed that particle exposed macrophages drive endothelial activation and that this mechanism is more potent than direct exposure of endothelial cells to diesel particles. The M1 associated cytokines, IL-12 and IFN-γ are increased in response to particulate matter. Although cigarette smoke shares similar characteristics to particulate pollution, exposure to cigarette smoke has been shown to skew macrophages towards an M2 phenotype.44 Treatment of polarised human monocyte derived M1 and M2 macrophages with diesel exhaust particles impairs expression of some M1 and M2 markers without overall inhibition of the polarisation process while increasing secretion of the M1 and M2 cytokines IL-8 and IL-10, respectively, via activation of Ahr and Nrf2.91 Although lung macrophages orchestrate the inflammatory responses induced by exposure to air pollutants resulting in adverse lung and cardiovascular effects, it remains to be determined which factors such as size and composition of particulate matter determine macrophage polarisation.
The ability to resist infectious diseases and mount protective immune responses declines with age; there is a high incidence of morbidity due to infectious diseases in the elderly population associated with RSV, viral influenza and pneumococcal pneumonia, compared with younger populations. An enhanced homoeostatic activation of the innate immune system in the elderly, in the absence of an immunological stimulus is referred to as ‘inflamm-aging’; this phenomenon is marked by elevated levels of tissue and circulating proinflammatory cytokines such as IL-1β, IL-6 and TNFα.92 Autophagy plays a critical role in the maintenance of macrophage homoeostasis and function but is reduced with increasing age. This results in macrophages with reduced phagocytosis and nitrite burst activity, while the inflammatory cytokine response is significantly increased.93 It has therefore been suggested that modulation of autophagy may preserve macrophage function during aging, therefore reducing the morbidity and mortality associated with inflamm-aging
Conclusion
It is evident that macrophages play a critical role in orchestrating the immune response to the inhaled environment and their manipulation presents an intriguing therapeutic avenue. Importantly, at least two pulmonary macrophage populations are known, AMs residing in the airways and IMs in the tissue compartment, respectively. Although each population likely shares various functions and roles with the other, each represents distinct lineages with specific origins, phenotypes and functions worthy of study and therapeutic exploitation. Indeed, a recent study in a hereditary pulmonary alveolar proteinosis model indicated that pulmonary macrophage transplantation was safe and well tolerated in mice.94 Multiple lines of investigation suggest that rather than focusing on the M1/M2 paradigm and studying in vitro differentiated monocytes, it may be more pertinent to garner an understanding of how these cells function in situ during chronic pulmonary diseases. Macrophages demonstrate a wide variety of roles and demonstrate a plastic nature; the challenge remains to apply the knowledge generated from mechanistic studies of macrophages towards directed therapies for the treatment of pulmonary disease.
Glossary
Macrophages were first described as ‘big eaters’ by their ability to phagocytose extracellular debris. Their ability to scavenge and degrade particulate matter from the local extracellular environment is critical in its role to maintain tissue homoeostasis.
Receptor mediated endocytosis
This process encompasses the ligation of specific receptors required for cell nutrients such as low-density lipoprotein, iron, vitamin A and vitamin D. Ligands are captured by numerous receptor-coated pits that upon binding promote an inward budding of the cellular membrane to produce vesicles. These vesicles fuse to form an endosome where the ligand can be released into the cytosol and receptors can be recycled to the cell surface.
Phagocytosis
This is the major process for the uptake and removal of microbial matter and particles greater than 5 µm in size. Particles are engulfed and taken up by the phagosome, a much larger endosome. Microbial uptake is initiated by the activation of pathogen-associated molecular patterns (PAMPS) by microbial ligands. The phagosome fuses with the lysosome where enzymes and peroxidases degrade the bacteria or particulate matter.
Efferocytosis
The role of efferocytosis is to recognise and remove apoptotic and necrotic cells before they release harmful and toxic substances to the local environment. Exposed phosphatidylserine or calreticulin is recognised by specific receptors on the macrophages. The cell membrane binds and engulfs the dying cell with it becoming encapsulated in an efferosome for degradation.
Autophagy
Autophagy is a constitutive intracellular degradation process that is necessary for cell survival particularly in response to nutrient stress. It plays a housekeeping role by clearing mitochondria, endoplasmic reticulum and peroxisomes and removing intracellular pathogens. Cytoplasmic constituents are encapsulated by a double membrane autophagosome, which are then delivered to the lysosome for degradation.
References
Footnotes
AJB, SAM contributed equally.
Twitter Follow Adam Byrne at @adbyrne
Contributors AJB, SAM and CML wrote, edited and reviewed the manuscript. LGG edited a revised version of the manuscript. CML supervised the project. CML and AJB are responsible for the overall content.
Funding Wellcome Trust (grant no. 087618/Z/08/Z).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.