Article Text

Abstract
Rationale Low-dose vitamin D supplementation is already recommended in older adults for prevention of fractures and falls, but clinical trials investigating whether higher doses could provide additional protection against acute respiratory infection (ARI) are lacking.
Objective To conduct a clinical trial of high-dose versus low-dose vitamin D3 supplementation for ARI prevention in residents of sheltered-accommodation housing blocks (‘schemes’) and their carers in London, UK.
Measurements and methods Fifty-four schemes (137 individual participants) were allocated to the active intervention (vitamin D3 2.4 mg once every 2 months +10 μg daily for residents, 3 mg once every 2 months for carers), and 54 schemes with 103 participants were allocated to control (placebo once every 2 months +vitamin D3 10 μg daily for residents, placebo once every 2 months for carers) for 1 year. Primary outcome was time to first ARI; secondary outcomes included time to first upper/lower respiratory infection (URI/LRI, analysed separately), and symptom duration.
Main results Inadequate vitamin D status was common at baseline: 220/240 (92%) participants had serum 25(OH)D concentration <75 nmol/L. The active intervention did not influence time to first ARI (adjusted HR (aHR) 1.18, 95% CI 0.80 to 1.74, p=0.42). When URI and LRI were analysed separately, allocation to the active intervention was associated with increased risk of URI (aHR 1.48, 95% CI 1.02 to 2.16, p=0.039) and increased duration of URI symptoms (median 7.0 vs 5.0 days for active vs control, adjusted ratio of geometric means 1.34, 95% CI 1.09 to 1.65, p=0.005), but not with altered risk or duration of LRI.
Conclusions Addition of intermittent bolus-dose vitamin D3 supplementation to a daily low-dose regimen did not influence risk of ARI in older adults and their carers, but was associated with increased risk and duration of URI.
Trial registration number clinicaltrials.gov NCT01069874.
- Respiratory Infection
Statistics from Altmetric.com
Key messages
What is the key question?
Does addition of intermittent bolus-dose vitamin D3 supplementation to a daily low-dose regimen enhance protection against acute respiratory infection in older adults and their carers?
What is the bottom line?
This intervention did not influence risk of acute respiratory infection in the study population, but it was associated with increased risk and duration of upper respiratory infection.
Why read on?
This clinical trial is the first study to evaluate the effect of vitamin D dosing interval on susceptibility to acute respiratory infection.
Introduction
Acute respiratory infections (ARIs) are the most common illnesses affecting humans. They have significant impact on health: upper respiratory infections (URIs, which primarily affect the respiratory tract above the vocal cords) are the most common cause of health-service consultation and productivity loss in industrialised countries,1 while lower respiratory infections (LRIs, primarily affecting the respiratory tract below the vocal cords) caused an estimated 2.81 million deaths worldwide in 2010.2 Existing prevention strategies focus on vaccination, but the effectiveness of this approach is limited as vaccines are lacking for many of the most common respiratory pathogens, and the immunogenicity of those in existence is impaired in older adults who are among the groups at highest risk.3 The development of alternative strategies to boost innate immune response to a broad range of pathogens could offer significant public health benefits.
Vitamin D supplementation represents one such strategy. The major circulating metabolite, 25-hydroxyvitamin D (25(OH)D) supports innate responses to both viral and bacterial respiratory pathogens.4 ,5 Older adults are at increased risk of suboptimal vitamin D status,6 ,7 and serum 25(OH)D concentrations <75 nmol/L have been reported to associate independently with increased risk of ARI.8 UK adults aged 65 years and older are advised to take a low-dose vitamin D supplement (10 µg=400 IU daily) for prevention of fractures and falls; no recommendation is made regarding vitamin D intake in younger adults who are widely perceived to be at low risk of vitamin D deficiency.9 Although daily lower-dose regimens have been reported to offer protection against URI in children,10 ,11 these regimens are insufficient to consistently elevate adults’ 25(OH)D concentrations above the 75 nmol/L threshold associated with optimum protection against ARI. A question, therefore, remains as to whether offering high-dose vitamin D supplementation to adults might offer additional protection against ARI as compared with daily low-dose supplementation.
We therefore conducted a pragmatic double-blind randomised placebo-controlled clinical trial among residents and carers in sheltered accommodation schemes in the UK to evaluate whether the addition of intermittent bolus dose vitamin D3 supplementation, administered once every 2 months for a period of 1 year, would offer superior protection against ARI as compared with standard of care (400 IU daily for residents and no supplementation for carers). A bolus dosing regimen was used to achieve rapid correction of vitamin D deficiency among participants in the intervention arm and to allow supervised administration of trial medication to maximise adherence. The trial was cluster-randomised, and carers were also enrolled, on the grounds that an intervention to prevent ARI in nursing home staff has previously been shown to reduce transmission of respiratory pathogens to those in their care.12
Methods
An abbreviated Methods section follows: further details are provided in online supplementary material.
Participants
Sheltered accommodation schemes in London, UK, were identified by searching the online directory at http://www.housingcare.org/ and assessed for their eligibility to host the trial: schemes offering care exclusively for clients with dementia, learning disability, mental health needs and alcohol or drug dependency were excluded. Housing associations responsible for potentially eligible sheltered accommodation schemes were then approached for permission to conduct the trial on their premises. Individual residents and their carers at sheltered accommodation schemes managed by participating housing associations were sent a letter inviting them to attend a screening visit. Principal exclusion criteria were presence of cognitive impairment or a communication problem precluding informed consent, medical record diagnosis of asthma or COPD and ingestion of a dietary supplement or prescribed therapy containing >10 μg (400 IU) vitamin D per day up to 2 months before first dose of study medication. Further details of eligibility criteria are provided in online supplementary material. The trial was approved by East London and The City Research Ethics Committee 1 (ref 09/H0703/112) and written informed consent was obtained from all participants before enrolment. The trial was registered at ClinicalTrials.gov (NCT01069874); the protocol is available from the corresponding author.
Procedures
Screening visit
Participants attending the screening visit completed the EuroQoL EQ-5D questionnaire.13 They also underwent a baseline clinical assessment, including measurement of height and weight and collection of blood sample for determination of serum concentrations of calcium, albumin and total 25(OH)D. A urine sample was collected from women of childbearing potential for a pregnancy test (SA Scientific, San Antonio, Texas, USA).
Run-in period
Participants fulfilling eligibility criteria then entered a run-in period of at least 2 weeks, during which they were asked to complete a study diary on a daily basis. This diary (see online supplementary figure S1) recorded the presence or absence of cough, cold or ‘flu symptoms for each day of participation in the trial. When symptoms were present, participants were also asked to record the severity of the following symptoms, scored from 0 (no symptoms) to 3 (symptoms severe enough to interfere with activity or sleep): headache, sneezing, rhinorrhoea, nasal congestion, sore throat, dyspnoea, wheeze, chest pain, cough, sputum production, sensation of fever or chilliness, myalgia and general malaise. The diary also recorded details of time off work (for carers only), healthcare use, medication use and out-of-pocket expenses incurred as a result of ARIs.
Randomisation
As soon as compliance with diary completion was demonstrated, and serum concentrations of corrected calcium and creatinine were available for at least one participant at a given sheltered accommodation scheme, this scheme was randomly assigned to active or control arms of the trial with a 1:1 ratio. Individual participants at randomised schemes then received one of the regimens detailed in table 1, according to (a) the allocation of the scheme at which they were enrolled, and (b) whether they were a resident or a carer at that scheme. All participants in the intervention arm received a total dose of 3 mg vitamin D3 over a 2-month period: for carers, this was given as a single bolus of 3 mg once every 2 months, while for residents, this was given as a daily dose of 10 µg plus a bolus dose of 2.4 mg once every 2 months. This regimen was designed to accommodate recommendations from the Department of Health that adults aged 65 years or more should receive a daily dose of 10 µg vitamin D in order to meet their reference nutrient intake.9 Details of the randomisation process are supplied in online supplementary material. Vitamin D3 content of a random sample of active medication was determined at the end of the study. Treatment allocation was concealed from participants and study staff. Randomised participants were invited to attend a subsequent study visit, at which the first dose of study medication was administered under direct supervision, and a new symptom diary was provided.
Active and control regimens
Follow-up
Participants were asked to complete study diaries daily for the 12 months of study participation. Each diary accommodated up to 12 weeks of data; participants completing follow-up filled six diaries in total. Five further bolus doses of study medication were administered once every 2 months following the first dose under direct supervision. Repeat blood samples were taken at 2 and 12 months, and serum was separated by centrifugation and frozen for subsequent assay of concentrations of 25(OH)D, albumin and calcium. Completion of the EQ5D questionnaire was repeated at 2, 6 and 12 months of follow-up. On completion of the 12-month visit, final diaries were collected, and participants were discharged from the study. Details of adverse events arising during the course of the trial and use of concomitant medications were recorded throughout. All study visits were conducted at participating sheltered accommodation schemes.
Data management and study definitions
All case report form and diary data were entered into a database in Microsoft Access 2010. Diary data were then imported into Stata, and episodes of ARI (categorised as either URI or LRI) were identified using algorithms based on the following definitions. URI was defined as (a) influenza-like illness as indicated by the presence of cough, feeling of fever/chilliness and muscle pain14 or (b) a cold, defined using the Jackson criteria.15 LRI was defined according to the Macfarlane criteria as follows. Each of five Macfarlane symptoms (cough, sputum production, dyspnoea, wheeze, chest discomfort/pain) was scored from 0 to 3, and an LRI was defined as presence of cough with symptom score at least one point over that recorded during the run-in period plus at least one other Macfarlane symptom scoring at least one point over that recorded during the run-in period.16 Further details of ARI definitions are supplied in online supplementary material.
Validation of ARI definition
In order to validate the diary definition for ARI, we performed paired nasopharyngeal and throat swabs on a subset of study participants during symptomatic events meeting ARI criteria and on occasions during which participants were asymptomatic.
Patients were sampled using flocked nasopharyngeal swabs (Copan Diagnostics, Murrieta, California, USA). Swabs were transferred to the laboratory in Universal Transport Medium (Copan Diagnostics), and tested for the presence of nucleic acids for ten respiratory pathogens (adenovirus; enterovirus; influenza A; influenza B; metapneumovirus; parainfluenza 1, 2 and 3; rhinovirus and respiratory syncytial virus) using real-time PCR.17
Sample size and statistical analysis
This trial was powered to detect a clinically significant difference in time to first ARI among participants enrolled in sheltered accommodation schemes allocated to active versus control arms of the trial (primary outcome). The proportion of the population experiencing at least one ARI per year is variously reported to be between 68% and 92%.1 ,16 ,18 Employing the Xie and Waksman formula for sample size estimation in clinical trials, with clustered survival times as the primary endpoint19 and assuming an average of three participants per unit, with intracluster coefficient of 0.05, equal numbers of units allocated to active and control arms of the study and 25% loss to follow-up of units, we calculated that a total of 108 units would need to be randomised to demonstrate a 20% reduction in proportion of participants experiencing at least one ARI in 1 year from 80% to 64%, with 80% power at the two-sided 5% significance level. This calculation was revised from the original power calculation, which indicated that we would need to randomise a total of 36 sheltered accommodation schemes, based on the assumption that 15 participants would be recruited in each scheme.
Prespecified secondary endpoints were the time to first URI and first LRI, the median duration of symptoms per episode, the proportion of participants experiencing at least one such episode, the rate of these episodes, the peak symptom score per episode, mean serum concentrations of 25(OH)D and corrected calcium at 2 and 12 months, unscheduled healthcare attendance for ARI, use of antibiotics and over-the-counter medications for treatment of ARI, quality of life as indicated by EQ5D scores; work absence (carers only), health economic outcomes (costs of ARI, quality-adjusted life years (QALY) and incremental net benefit over 1 year) and incidence of adverse events. Prespecified subgroup analyses were conducted to determine whether the effect of vitamin D3 supplementation on risk of ARI, URI and LRI was modified by the type of participant (resident vs carer).
Analyses were performed using Stata/IC (V.12.1, 2012, and V.13, 2013), GraphPad Prism (V.4.03, 2005) and R (V.3.0.2, 2013) software packages. Analysis was by intention-to-treat (ITT), and significance was tested at the 5% level. Time-to-event outcomes were analysed using Cox regression adjusted for minimisation variables (level of care, size of scheme and season of randomisation) and participant study group (resident vs carer), allowing for a shared frailty within the same unit with frailty following a gamma distribution. Effects of allocation on time-to-event outcomes are presented as HRs, with the numerator being the hazard or chance of the outcome occurring in the intervention arm and the denominator being the hazard or chance of the outcome occurring in the control arm; thus, a HR>1 represents an increased risk of the outcome occurring in the intervention arm and vice versa. Further details of statistical analyses, including health economic analyses, are provided in online supplementary material.
Laboratory analyses
Serum concentrations of 25(OH)D2 and 25(OH)D3 were determined by isotope-dilution liquid chromatography–tandem mass spectrometry20 and summed to give values for total 25(OH)D concentration. Sensitivity for this assay was 10 nmol/L. Albumin and total serum calcium concentrations were determined using an Architect ci8200 analyser (Abbott Diagnostics, Chicago, Illinois, USA). Calcium concentration was corrected for serum albumin concentration using the formula: corrected calcium (mmol/L)=total calcium (mmol/L)+0.02×(40−albumin (g/L)). Vitamin D3 content of active medication was determined by high-performance liquid chromatography.
Results
Two hundred and seventy-seven adults, resident or working in 116 sheltered accommodation schemes were assessed for eligibility to participate in the trial between 29 March 2010 and 22 March 2012: 29 were ineligible to participate, and 8 were eligible, but declined randomisation. The remaining 240 participants were either residents or carers at 1 of 108 schemes, which were then randomised to active versus control arms of the trial in equal numbers: 54 schemes with a total of 137 individual participants (115 residents, 22 carers) were assigned to the active arm of the trial, and 54 schemes with a total of 103 individual participants (79 residents, 24 carers) were assigned to the control arm. All participants received at least one dose of study medication, and were included in the ITT analysis (figure 1). Clinical and demographic characteristics of randomised participants were comparable for active versus control groups (table 2). The majority of participants (220/240, 92%) had inadequate vitamin D status (serum 25(OH)D <75 nmol/L) at baseline. The trial ended on the date of the final study visit of the final participant undergoing follow-up.
Baseline characteristics by allocation
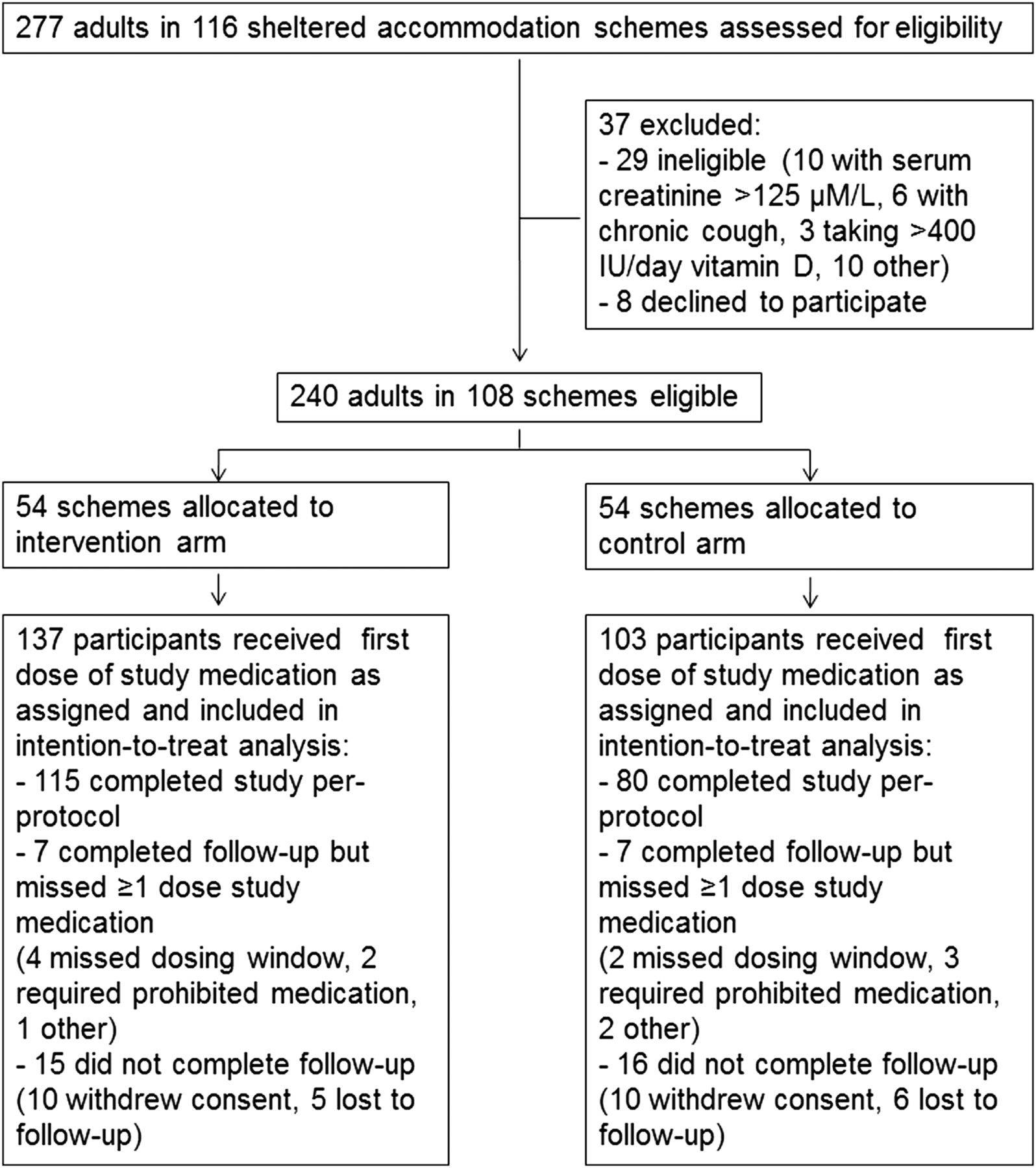
Trial profile.
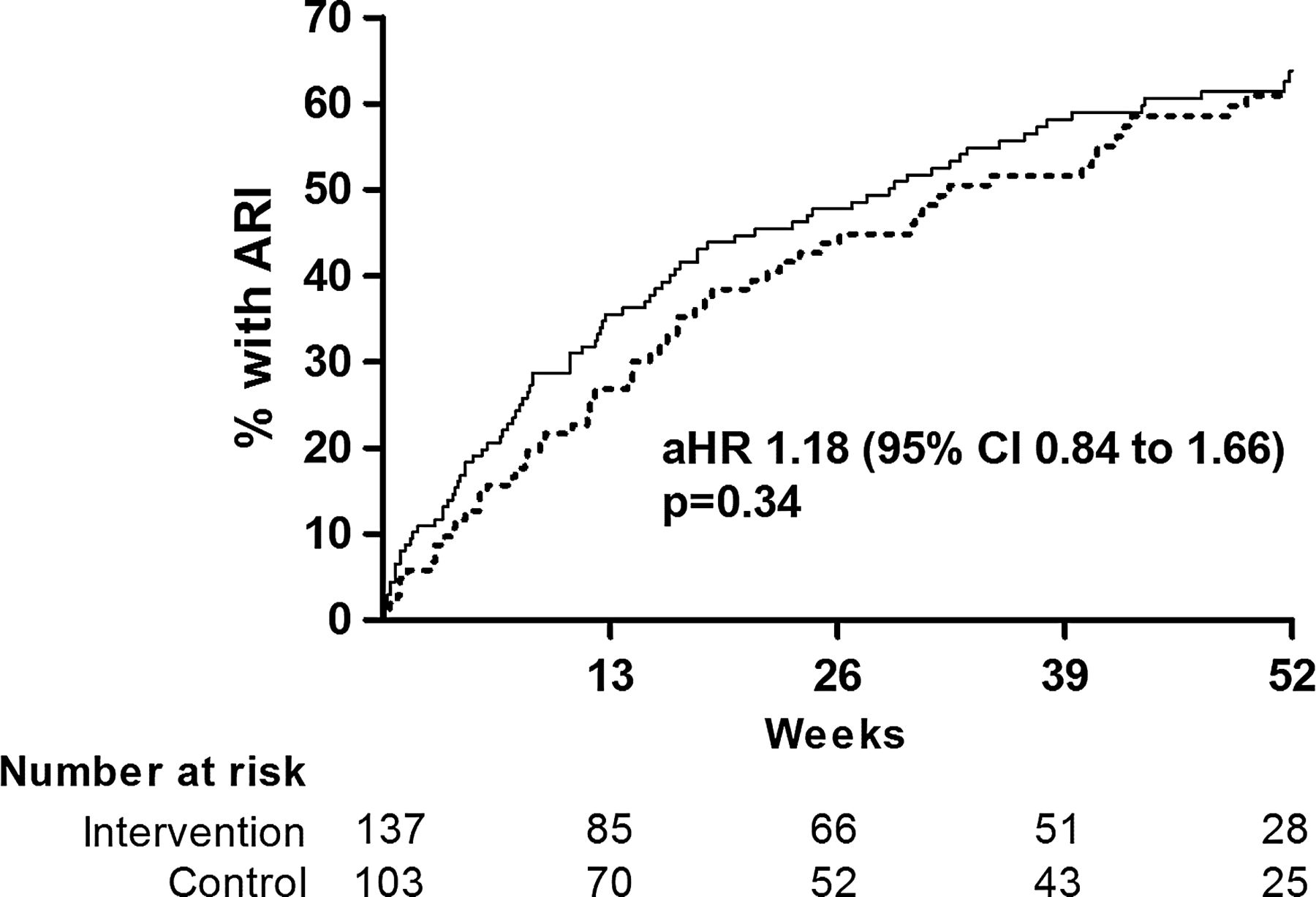
Allocation to the active versus the control arm of the trial did not significantly influence time to first ARI (adjusted HR (aHR) 1.18, 95% CI 0.84 to 1.66, p=0.34, figure 2), the proportion of participants experiencing at least one ARI (adjusted OR (aOR) 1.21, 95% CI 0.68 to 2.15, p=0.52), the rate of ARI per participant year (adjusted incidence rate ratio 1.15, 95% CI 0.84 to 1.58, p=0.37) or median ARI duration (adjusted ratio of geometric means 1.17, 95% CI 0.92 to 1.49, p=0.21, table 3). However, when URI and LRI were analysed separately—as prespecified in the protocol and the analysis plan—allocation to the active arm of the trial was associated with increased risk of URI (aHR 1.48, 95% CI 1.02 to 2.16, p=0.039, figure 3), and increased median duration of URI symptoms (7 vs 5 days for active vs control, adjusted ratio of geometric means 1.34, 95% CI 1.09 to 1.65, p=0.005). Allocation did not have a differential effect on time to first URI fulfilling Jackson criteria for cold (aHR 1.40, 95% CI 0.95 to 2.07, p=0.09) versus time to first URI fulfilling criteria for influenza-like illness (aHR 1.38, 95% CI 0.73 to 2.61, p=0.32). No effect of allocation was seen on LRI outcomes (table 3). Vitamin D status improved in both the active arm (40.4 nmol/L mean increase in serum 25(OH)D at 12 months vs baseline, 95% CI for difference 36.0 to 44.7 nmol/L, p<0.001) and the control arm (14.9 nmol/L mean increase in serum 25(OH)D at 12 months vs baseline, 95% CI for difference 10.5 to 19.3 nmol/L, p<0.001). At 12 months, mean serum 25(OH)D was higher in the active versus the control arm of the trial (adjusted mean difference 25.7 nmol/L, 95% CI 20.6 to 30.7 nmol/L, p<0.001), but mean serum corrected calcium concentration at 12 months was not significantly different between arms (p=0.21, table 4). A random sample of active medication assayed at the end of the study was found to contain 99.2% of its original vitamin D3 content.
Respiratory outcomes by allocation
Biochemical outcomes by allocation
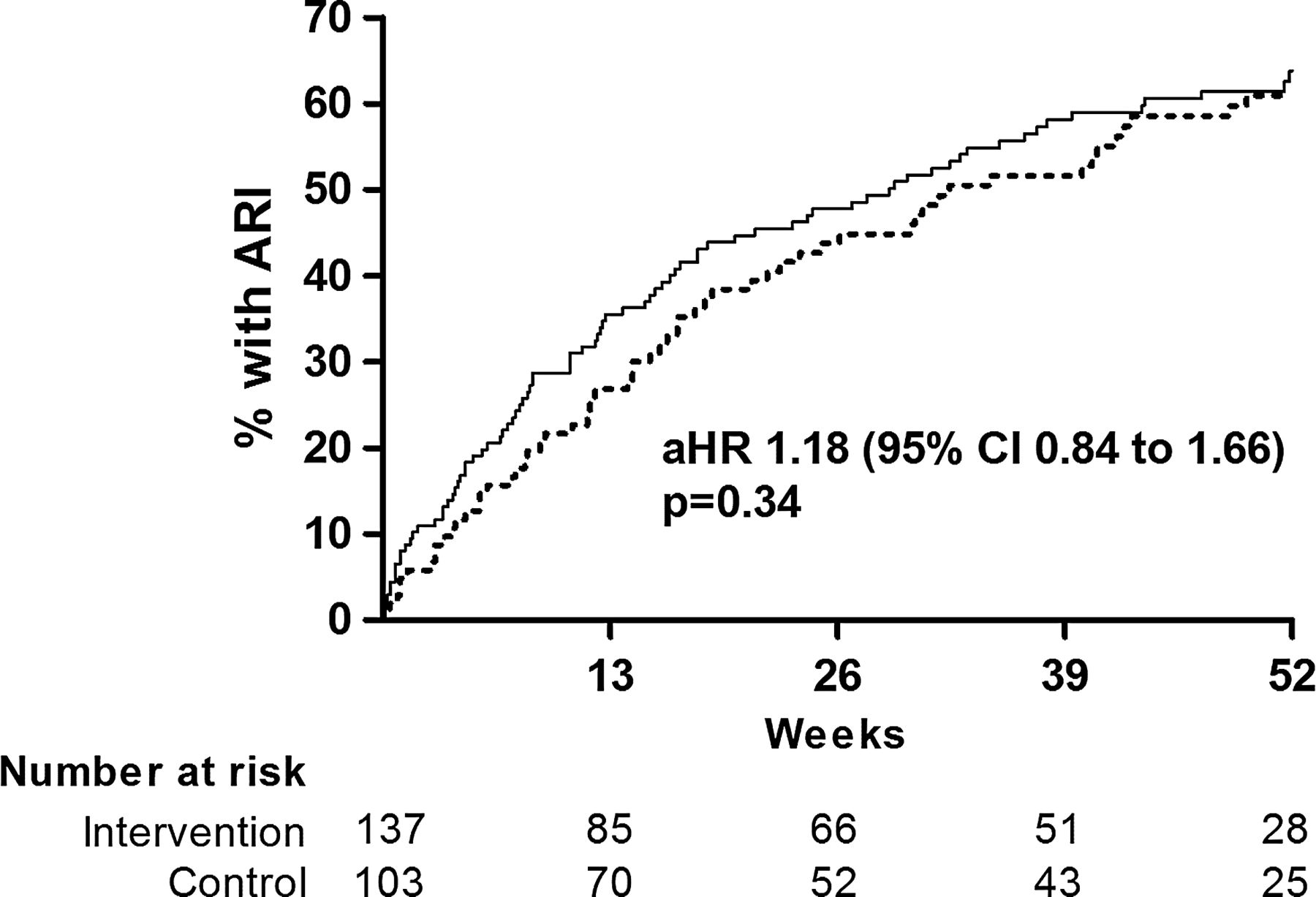
Time to first ARI by allocation. Numbers of participants yet to experience ARI (number at risk) at 0, 13, 26, 39 and 52 weeks are shown. aHR, adjusted HR; ARI, acute respiratory infection. Solid line, active arm; dotted line, control arm.

{kind=link}
{kind=link}
{kind=link}
Time to first URI by allocation. Numbers of participants yet to experience URI (number at risk) at 0, 13, 26, 39 and 52 weeks are shown. aHR, adjusted HR; URI, upper respiratory infection. Solid line, active arm; dotted line, control arm.
No effect of allocation was seen on unscheduled healthcare attendance or use of antibiotics or over-the-counter medications for ARI (see online supplementary table S1) or on quality-of-life outcomes as evaluated with the EQ5D questionnaire (see online supplementary table S2). Allocation did not influence risk of work absence due to ARI among carers (see online supplementary table S3) or health economic outcomes (ARI-associated costs, QALY and incremental net benefit; see online supplementary table S4). The cost-effectiveness acceptability curve displayed in online supplementary figure S2 shows that the probability that the active intervention is cost-effective for prevention of ARI is less than 60% at a realistic willingness to pay (£20 000) for a QALY gain.
In order to dissect out effects of the distinct study regimens administered to residents versus carers, we conducted a prespecified subgroup analysis to determine the influence of allocation on time to ARI, URI and LRI within each of these groups (see online supplementary table S5). Allocation to the intervention arm of the trial was associated with increased risk of URI among residents (aHR 1.58, 95% CI 1.02 to 2.43, p=0.039), but not among carers (aHR 1.24, 95% CI 0.47 to 3.26, p=0.67). No other statistically significant effect of allocation was seen for other outcomes investigated in this subgroup analysis.
Forty-seven serious adverse events were reported in 39/240 participants receiving at least one dose of study medication; none of these were attributed to study medication, and no participant died during the study (see online supplementary table S6). A total of 1132 non-serious adverse events were reported in 221/240 participants: other than a trend towards more self-reported ARI in active versus control arms (267 vs 238, respectively), these were equally distributed between study arms (see online supplementary table S7).
To validate the diary definition for ARI, paired nasopharyngeal and throat swabs were taken in study participants during 21 symptomatic events meeting ARI criteria, and on 145 occasions during which participants were asymptomatic, and tested for the presence of ten respiratory pathogens as described in Methods. Symptomatic events were strongly associated with detection of at least one of the pathogens above (detected in 11/21 symptomatic episodes vs 7/145 patients who were asymptomatic, p<0.0001). Pathogens detected during symptomatic episodes were rhinovirus alone (n=5), influenza A (n=2), enterovirus alone, influenza B, parainfluenza 3 (n=1 each) and mixed rhinovirus/enterovirus infection (n=1).
Discussion
We present results of the first randomised controlled trial to compare the efficacy of intermittent bolus-dose versus low-dose daily vitamin D3 supplementation for the prevention of ARI. In a population with high baseline prevalence of vitamin D deficiency, addition of intermittent bolus-dose vitamin D3 supplementation to standard of care (400 IU/day for older adults, nil for younger adults) elevated vitamin D status, but did not influence risk of ARI. However, when URI and LRI were analysed separately, allocation to the active intervention was found to increase risk of URI and duration of URI symptoms.
One potential explanation of this finding is that higher serum 25(OH)D levels achieved in the intervention arm might have impaired immunity to pathogens causing URI. A U-shaped relationship between serum 25(OH)D and risk of TB has previously been reported, with very low and very high levels both associated with increased risk of disease.21 However, observational studies reporting that serum 25(OH)D levels >75 nmol/L22 or >96 nmol/L23 associate with protection against ARI do not support this interpretation. Alternatively, it may be that the intermittent bolus dosing regimen we used in the active intervention is less effective than daily dosing at inducing protective immunity to URI. Administration of vitamin D using large intermittent oral boluses has been hypothesised to dysregulate vitamin D metabolism in extrarenal tissues by causing inappropriately low 1-alpha-hydroxylase activity coupled with inappropriately high 24-hydroxylase activity, which may limit local concentrations of the active vitamin D metabolite 1,25-dihydroxyvitamin D.24 It has also been suggested that ‘parent vitamin D3’ (cholecalciferol) may itself play an important physiological role:25 since the half-life of this compound is only ∼24 h, daily dosing with 400 IU would have caused sustained elevation of cholecalciferol levels, whereas dosing at 2-month intervals would not have. In this context, it is interesting to note that relatively low daily doses of vitamin D have been shown to offer protection against URI despite causing relatively modest increments in 25(OH)D,10 ,26 and that trials of intermittent bolus dosing regimens for URI prevention have tended to be null.27–29
Our study has several strengths. Inadequate vitamin D status was very common among the study population at baseline, and adherence to bolus doses of study medication was high as administration of all such doses was directly supervised by study staff. We collected detailed prospective data on outcomes by using a PCR-validated case definition. This allowed us to detect potential effects of the intervention on episodes that did not come to medical attention, and to determine the influence of allocation on symptom severity and duration as well as incidence of ARI. We also collected data on quality of life and ARI-associated costs, allowing us to conduct a health economic evaluation of the intervention.
Our trial also has some limitations. By using a cluster-randomised design, we hoped to demonstrate benefits of achieving ‘herd immunity’ to pathogens causing ARI by enrolling the majority of residents and carers in a given scheme and allocating them all to the same intervention. The proportion of residents/carers enrolling in the trial at each scheme was lower than expected, however, and this precluded detection of such effects. The total number of carers enrolled was small; null effects of the intervention observed in this subgroup could, therefore, be due to lack of power. Sampling of vitamin D status was limited to 2-month and 12-month time points, so the 25(OH)D concentrations measured represent ‘trough’ values only; the 25(OH)D response to bolus-dose supplementation would have been better characterised if vitamin D status had additionally been measured at 3–7 days post dose when it would have been expected to peak.
In summary, we have shown that addition of intermittent bolus-dose vitamin D supplementation to standard of care elevated vitamin D status among residents and their carers in sheltered accommodation units, but increased risk of URI. Our findings suggest that intermittent bolus dose vitamin D supplementation is less effective than daily supplementation for prevention of URI. The influence of dosing interval on immune function and susceptibility to ARI should be evaluated in clinical trials comparing regimens that deliver the same total dose of vitamin D administered in daily versus intermittent doses.
Acknowledgments
We thank the members of our data monitoring committee, Guy E Thwaites (Chair), Brenda E Jones and Tuan Q Phung. We also thank housing associations and scheme managers responsible for study sites, pharmacy staff at the Royal London Hospital for overseeing the dispensing of study medication and all the people who participated in the trial.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors ARM, YH, NCB, SME and CJG contributed to study design. ARM, YH, KDW, MP, NS, ZE, ZB, AS, AK and DAJ participated in implementation of the trial. CLG, PMT, TRV, MR and DC developed and ran laboratory assays. DM validated diary records of health economic outcomes. ARM and RLH contributed to data analysis. ZS performed health economic analysis. ARM wrote the first draft of the manuscript; all other authors critically reviewed it, and approved the final version.
Funding This is a summary of independent research funded by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research Programme (Reference Number RP-PG-0407-10398). The views expressed are those of the authors, and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests None declared.
Ethics approval East London and The City Research Ethics Committee 1, London, UK.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data from the study will be made publicly available in a data repository on publication of this trial report.