Article Text

Abstract
Background Airway macrophage (AM) phagocytosis is impaired in severe asthma. Prostaglandin (PG) E2 and D2 are increased in severe asthma and suppress AM phagocytic function in vitro. In this study, we sought evidence for PG-mediated impairment of phagocytosis of inhalable carbonaceous particulate matter (PM) by AM in children with severe asthma compared with mild asthmatics and healthy controls.
Methods AM were obtained from children with asthma and healthy controls using induced sputum. AM carbon area (μm2) was assessed by image analysis. In a subgroup of asthmatics, urinary PGE2 and PGD2 metabolites were measured by high-performance liquid chromatography, and PM exposure at the home address was modelled. Phagocytosis of PM by human monocyte-derived macrophages and rat AM was assessed in vitro by image analysis.
Results AM carbon was 51% lower in children with moderate-to-severe asthma (n=36) compared with mild asthmatics (n=12, p<0.01) and healthy controls (n=47, p<0.01). There was no association between modelled PM exposure and AM carbon in 33 asthmatics who had a urine sample, but there was an inverse association between AM carbon and urinary metabolites of PGE2 and D2 (n=33, rs=−0.40, p<0.05, and rs=−0.44, p<0.01). PGE2 10−6 M, but not PGD2 10−6 M, suppressed phagocytosis of PM10 by human macrophages in vitro (p<0.05 vs control). PGE2 10−6 M also suppressed phagocytosis of PM10 by rat AM in vitro (p<0.01 vs control).
Conclusions Phagocytosis of inhaled carbonaceous PM by AMs is impaired in severe asthma. PGE2 may contribute to impaired AM phagocytic function in severe asthma.
- Macrophage Biology
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is there evidence of impaired phagocytosis of inhaled carbonaceous particulate matter (PM) in children with severe asthma, and is there a role for prostaglandin (PG) E2 and D2 in mediating suppression of phagocytosis?
What is the bottom line?
-
The amount of carbon in airway macrophages (AM) from children with severe asthma is 51% lower than healthy controls and mild asthmatics, urinary markers of PGE2 and PGD2 are increased in severe asthma, and PGE2 suppresses phagocytosis of PM by human macrophages in vitro.
Why read on?
-
AM carbon is lower in severe asthma, and PGE2 suppresses AM phagocytosis of PM, results that lead to testable questions; what are the effects of inhaled PM that evades normal AM clearance, and does blocking PGE2 release improve AM phagocytic capacity?
Introduction
Studies in children and adults suggest that the capacity of airway macrophages (AMs) to phagocytose inhaled material is impaired in severe asthma. First, Fitzpatrick et al1 reported a 50% reduction of phagocytosis of Staphylococcus aureus by AM from children with poorly controlled asthma. Second, Alexis et al2 reported that phagocytosis of opsonised yeast by AM is impaired in adults with eosinophilic asthma compared with healthy controls. Third, Huynh et al3 reported impaired phagocytosis of apoptotic human T cells by AM from adults with severe asthma compared with either healthy controls or adults with less-severe asthma. To date, the mechanism for impaired AM phagocytosis in asthma is unclear, but severe asthma is associated with increased airway levels of eicosanoids that suppress macrophage phagocytosis in vitro. For example, prostaglandin E2 (PGE2) is increased in induced sputum from adults with eosinophilic asthma4 ,5 and inhibits the phagocytosis of opsonised Escherichia coli and sheep red blood cells by rat AM in vitro.6 Furthermore, PGE2 inhibits the phagocytosis of sheep red blood cells by murine AM in vitro,7 and PGE2, and to a lesser extent PGD2, inhibits phagocytosis of apoptotic neutrophils by human macrophages in vitro.8 To date, it is unknown whether PGE2 and PGD2 suppress macrophage phagocytosis of fossil fuel-derived particulate matter (PM) in vitro.
Carbonaceous PM is phagocytosed in a dose-dependent manner by AM.9 ,10 Studies in animals and humans suggest that the amount of PM in AM is a valid marker of chronic exposure to carbonaceous PM from fossil fuel combustion. For example, in rats, the amount of carbon in AM reflects exposure to diesel PM over a 13-week period,11 and in human adults the amount of carbon in AM is most strongly associated with the modelled mean exposure to inhalable PM (PM <10 microns in aerodynamic diameter; PM10) over the previous 6 months.12 We, and other groups, have therefore used the area of carbon in AM (AM carbon) to assess the health effects of chronic exposure to PM in children and adults.13–16 The utility of AM carbon as a marker of personal exposure to PM is based on the assumption that the phagocytic capacity of AM is broadly similar between individuals. However, in a previous small pilot study, we unexpectedly found low to absent AM carbon in a small number of children with severe eosinophilic asthma and speculated that phagocytosis of AM carbon in vivo is impaired in severe asthma.14 In the present study, we sought to establish definitive evidence for (i) lower AM carbon in children with severe asthma compared with children with either mild asthma or healthy controls, and (ii) a role for PGE2 and PGD2 in suppressing phagocytosis.
Methods
Participants
Asthmatic children were recruited from the Royal London Children's Hospital, the University Hospitals of Leicester, the Royal Brompton Hospital (London) and schools in east London. The major source of carbonaceous PM exposure in children in these urban areas is fossil fuel-derived PM. Current asthma control was established from a review of the clinical notes or a parent-completed International Study of Asthma and Allergies in Childhood questionnaire.17 Asthma severity was categorised by British Thoracic Society/Scottish Intercollegiate Guidelines Network (BTS/SIGN) criteria. Children with asthma at BTS steps 1–2 were classified as ‘mild’, and BTS steps 3–5 classified as ‘moderate-to-severe’.18 Healthy children were recruited from east London schools and from siblings of children attending asthma clinics. A subgroup of asthmatic children recruited at the Royal London Children's Hospital also had a urine sample obtained and exposure of their home to PM modelled using (i) the London Air Quality Toolkit (LAQT) and (ii) the home to main road distance. The LAQT is an established emissions dispersion model capable of producing annual mean pollutant concentrations at a resolution of 20 m×20 m.19 The LAQT models the mean exposure of children's home addresses to PM with mean aerodynamic diameter <2.5 μm (PM2.5), for annual, 7-day and 24 h PM2.5 prior to sputum induction (see online supplement). Children living in homes within 50 m of a main road were considered to have increased exposure to traffic-derived PM. Written informed assent and consent was obtained from children and parents (Research Ethics Committees approval reference; 08/H0704/139, 11/LO/1732 and 09/H0403/92).
Spirometry and induced sputum
Spirometry was performed according to standard criteria20 using a Microlab spirometer (Care Fusion, Kent, UK). Post bronchodilator lung function was measured 15 min after 400 μg inhaled salbutamol via a metered dose inhaler and large volume spacer. SD scores (z scores) for FEV1 and FVC were calculated using Global Lung Initiative software.21 Children with asthma underwent sputum induction during a clinician-assessed period of stability. Induced sputum was not done within 1 month of an asthma exacerbation requiring oral corticosteroids. Sputum induction and processing was done as previously described14 using nebulised 4.5% saline via a Multisonic Profi nebuliser (Schill, Germany) (see online supplement).
AM carbon
The area of carbon in AM was determined as previously reported.14 Briefly, digital images of 50 randomly selected AM from each child were obtained at ×100 magnification, and the mean area (μm2) of carbon determined. Analysis was blinded to asthma status. Additional details are given in the online supplement.
Urinary PGE2 and PGD2 metabolites
Urine was obtained from children at sputum induction, transported on ice and stored at −80°C within 1 h of collection. Analysis was by high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS).22 Negative ionisation was used to detect 13,14-dihydro-15-keto-tetranor-PGE2 (-tetranor-PGE2) and 13,14-dihydro-15-keto-tetranor-PGD2 (-tetranor-PGD2). Metabolite concentrations were expressed in proportion to urinary creatinine (see online supplement).
Phagocytosis of PM10 in vitro
The effect of PGE2 and PGD2 on the phagocytosis of urban PM10 by human monocyte-derived macrophages and rat AM was assessed using a modification of the in vitro assay described by Aronoff et al6 (see online supplement). The aim was to first assess the effect of PGE2 and PGD2 on human macrophages at the optimal suppressive concentration (10−6 M) described by Aronoff et al,6 then confirm any positive findings using rat AM. Briefly, either human monocyte-derived macrophages from healthy adult donors or rat AM were adhered to bovine serum albumin-coated coverslips and cultured with PM10 (10 µg/mL) collected from urban filters in Leicester city centre with or without PGE2/PGD2 (Sigma-Aldrich, St Louis, Missouri, USA) for 1 h.6 Phagocytosis of PM10 was determined by image analysis of 50 cells per well by an operator blinded to exposure status (described above). Experiments were done on at least six separate occasions.
Statistical analysis
Data are presented as median (IQR). Correlations were done by Spearman's rank correlation (rs) and proportions compared by χ2 test. Data between two groups were compared by either Mann–Whitney test (unpaired) or Wilcoxon matched-pairs signed-rank test (paired). Comparisons between more than two groups were done by Kruskal–Wallis test and Dunn's multiple comparison test. Analyses were performed using Prism 5.00 for Windows (GraphPad Software, California, USA). Results were considered significant at p<0.05.
Results
AM carbon and induced sputum eosinophil differential count (figure 1) were determined in 47 healthy controls, 13 children with asthma at BTS steps 1–2 and 36 children with asthma at BTS steps 3–5 (figure 2, table 1). Subject numbers were determined by the study duration (October 2011–December 2012, see online supplementary figure S1). For asthmatic and healthy children recruited at the Royal London Children's Hospital and in east London schools, 195 children were approached, 129 consented to undergo a sputum induction and a technically acceptable induced sputum (IS) sample was obtained from 72 (56%). One asthmatic child with sufficient number of cells for AM carbon had insufficient leucocytes for an induced sputum differential count. All 33 asthmatic children recruited at the Royal London Children's Hospital provided a urine sample for PG analysis. At the University Hospitals of Leicester, 20 children with asthma and 58 healthy controls were approached for IS, 20 asthmatic children and 17 controls agreed to undergo IS. Ten randomly sampled slides from each group were assessed for AM carbon, and AM carbon determined in eight asthmatics and eight controls (see online supplementary figure S1). At the Royal Brompton Hospital, 65 children with asthma were approached for IS, 55 agreed to undergo IS. Eight randomly sampled slides were assessed and AM carbon determined in 8/8 (see online supplementary figure S1). For analysis, data from all three sites were pooled.
Demographics and airway macrophage carbon in healthy controls and children with asthma

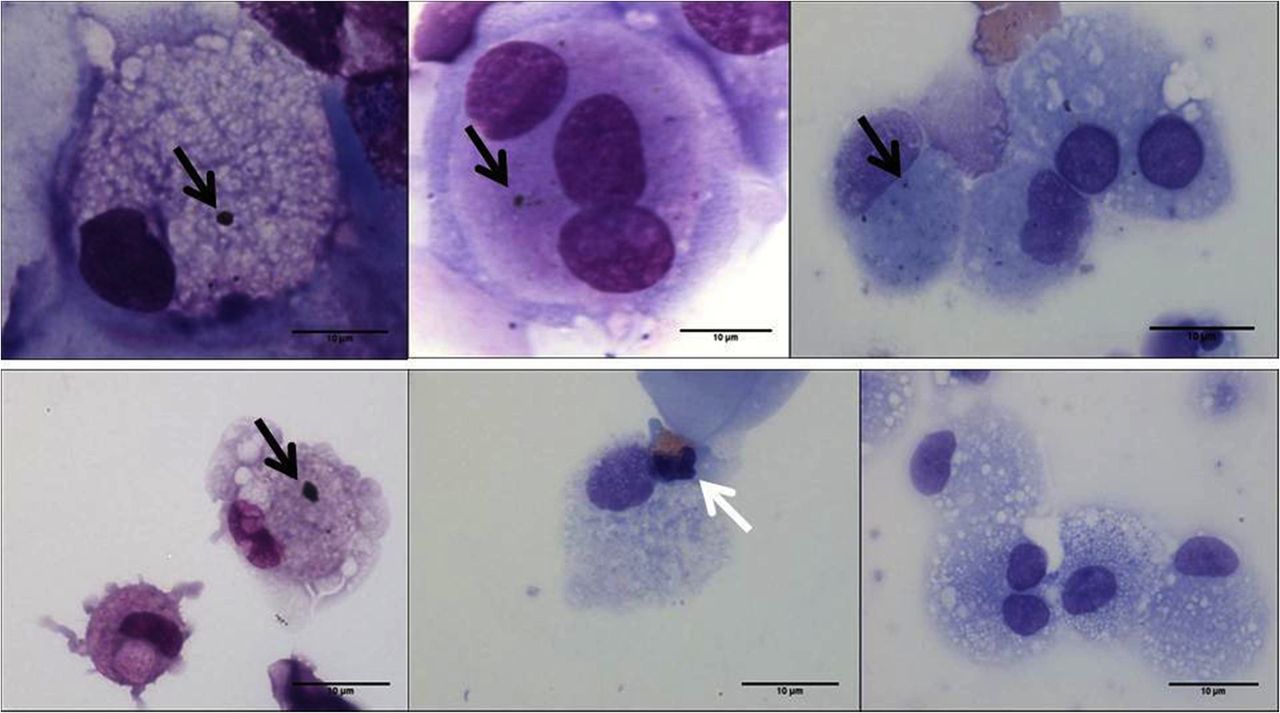
Induced sputum airway macrophages (AM) from children showing phagocytosed inhaled carbon. The black arrows show carbon in AM from healthy children (top row) and from children with moderate-to-severe asthma (bottom row). The white arrow indicates an eosinophil. Images at ×100, bar=10 µm.

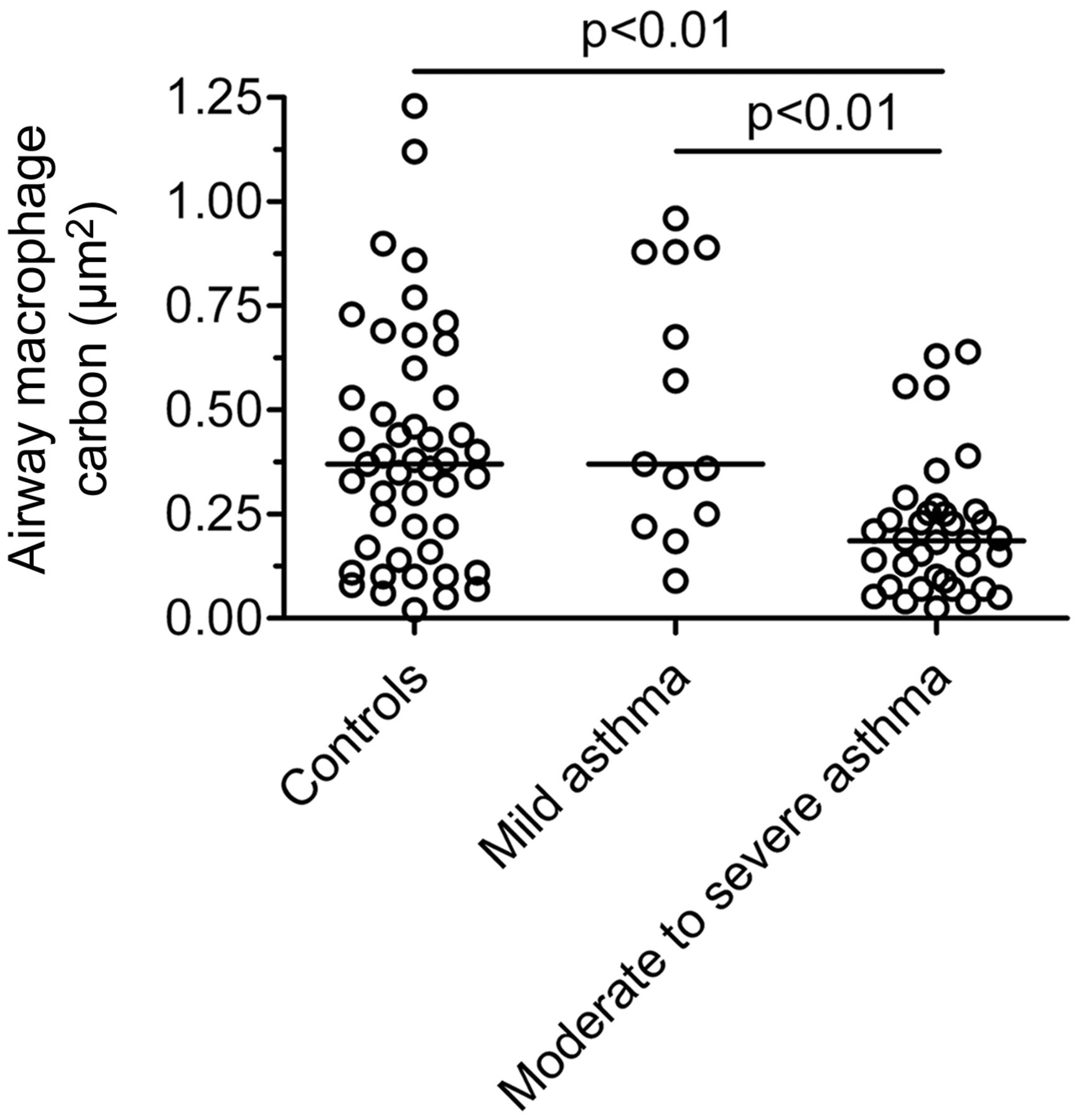
Comparison of airway macrophage (AM) carbon between children with mild asthma (British Thoracic Society (BTS) steps 1–2), moderate-to-severe asthma (BTS steps 3–5) and healthy controls. AM carbon was calculated from 50 AM per child and is expressed as the mean area of carbon per AM (μm2). Comparison is by Kruskal–Wallis test and Dunn's multiple comparisons test. Bar represents median. Children with moderate-to-severe asthma have lower AM black carbon compared with both healthy controls and mild asthmatics. There is no significant difference between healthy controls and mild asthmatics.
Compared with healthy controls, children with moderate-to-severe asthma (BTS steps 3–5), but not those with mild asthma (BTS steps 1–2), had lower postbronchodilator FEV1 z score and increased induced sputum eosinophils (table 1). In healthy controls, there was no association between AM carbon and age, and no difference in AM carbon between males and females. There was also no significant difference in AM carbon when stratified by site of recruitment in either control or asthmatic groups.
Asthma and AM carbon
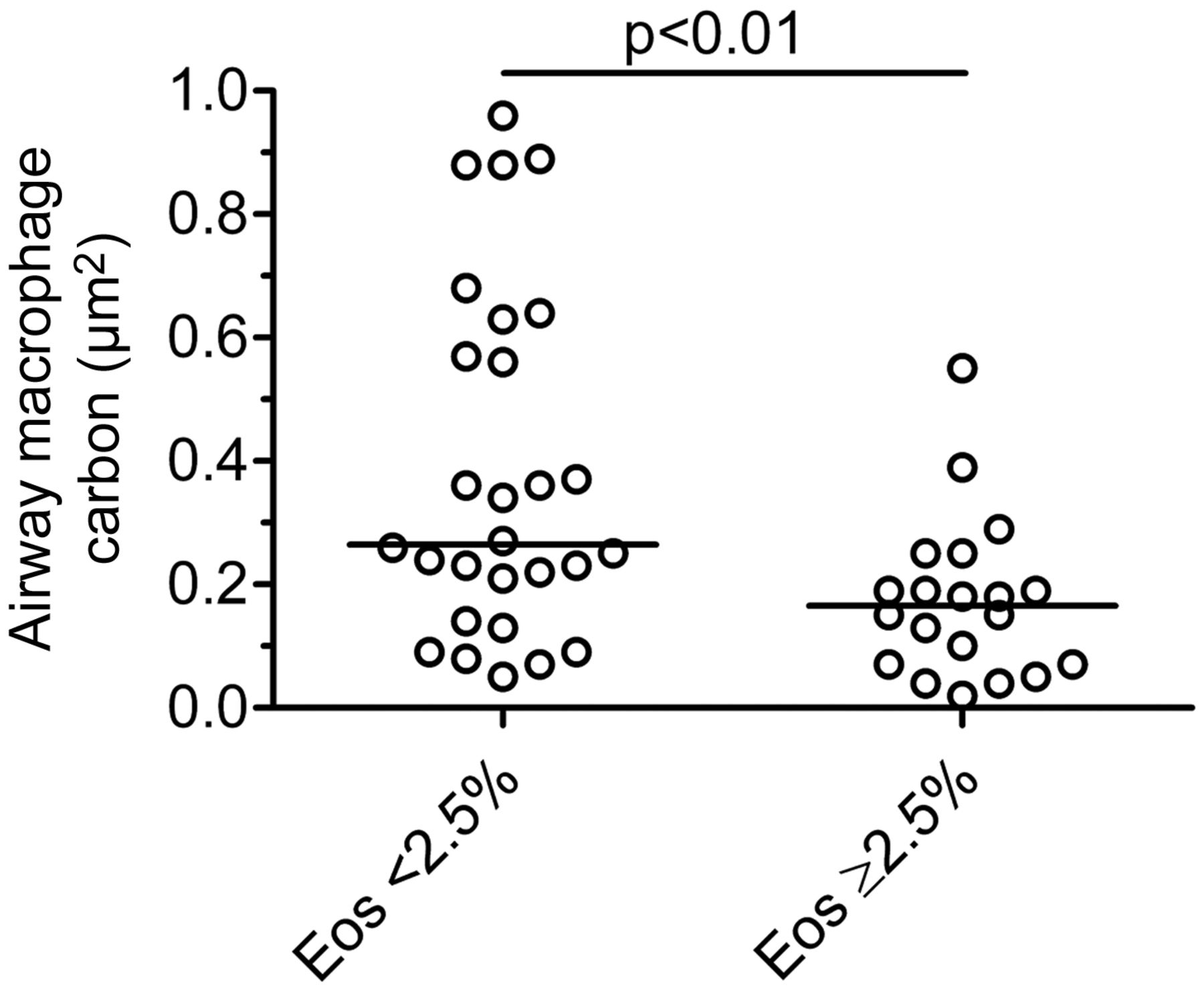
Asthmatic children with moderate-to-severe asthma had 51% less AM carbon compared with both healthy controls (p<0.01) and mild asthmatics (p<0.01, table 1, figure 2). There was no difference in AM carbon between healthy controls and children with mild asthma (table 1, figure 2). In asthmatics (n=49), there was an inverse association between AM carbon and sputum eosinophils (%) (rs=−0.32, p<0.05), but not with sputum neutrophils (%) (p=0.14). AM carbon was therefore lower in asthmatic children with sputum eosinophilia defined as ≥2.5% (0.16 vs 0.25 μm2, p<0.01, figure 3). AM carbon in asthmatics was also inversely associated with daily inhaled corticosteroid dose (rs=−0.31, p<0.05) and age (rs=−0.44, p<0.01).

Comparison of airway macrophage (AM) carbon between children with eosinophilic (≥2.5%, n=20) and non-eosinophilic (<2.5%, n=28) asthma. Eosinophil (Eos) differential count is from 400 induced sputum leucocytes per child. AM carbon is lower in children with eosinophilic asthma (p<0.01, Mann–Whitney test). Bar represents median.
Urinary PG metabolites and AM carbon
In the 33 asthmatic children who had a urine sample taken for PG analysis, exposure to air pollution at the home address was determined by the LAQT air pollution model for 23/33, and the home–road distance assessed in 33/33. There was no difference in modelled annual mean PM2.5, 7-day PM2.5 or 24 h PM2.5 between mild and moderate-to-severe asthmatics (table 2), and no difference in the proportion of homes within 50 m of a main road between the two asthmatic groups (table 2). There was no association between modelled PM exposure and AM carbon in 33 asthmatics who had a urine sample. Urinary PG metabolites (13,14-dihydro-15-keto-tetranor-PGE2 and 13,14-dihydro-15-keto-tetranor-PGD2) were highly correlated (rs=0.89, p<0.001), and both metabolites were increased in moderate-to-severe asthma (p<0.05, p<0.01 vs mild asthmatics for -tetranor-PGE2 and -tetranor-PGD2, figure 4, table 2). AM carbon was inversely associated with urinary metabolites of PGE2 (rs=−0.40, p<0.05) and PGD2 (rs=−0.44, p<0.01). There was an association between sputum eosinophils and urinary PGE2 and PGD2 metabolites (rs=0.35, p<0.05, and rs=0.39, p<0.05, respectively). Compatible with results from all children, AM carbon in the subgroup with a urine sample was lower in moderate-to-severe asthmatics compared with mild asthmatics (p<0.01, table 2, see online supplementary figure S2).
Prostaglandin E2 and D2 metabolites and modelled exposure to air pollution in asthmatic children in whom a urine sample was obtained

Effect of asthma severity on urinary metabolites of (A) prostaglandin E2 (PGE2) and (B) PGD2 in the subgroup of 33 asthmatic children where a urine sample was obtained. Urinary 13,14-dihydro-15-keto-tetranor-PGE2 and 13,14-dihydro-15-keto-tetranor-PGD2 (-tetranor-PGE2/D2) is higher in moderate-to-severe asthmatics compared with mild asthmatics. Urinary metabolites of PGE2 and PGD2 were measured using high-performance liquid chromatography–tandem mass spectrometry. Comparison by Mann–Whitney test. Bar represents median.
Phagocytosis of PM10 in vitro
Phagocytosis of PM10 by human monocyte-derived macrophages was suppressed by PGE2 10−6 M (p<0.05 vs control, figure 5A), but not by PGD2 10−6 M (figure 5B). PGE2 at 10−6 M also suppressed phagocytosis of PM10 by rat AM (p<0.05, figure 6).

Effect of (A) prostaglandin E2 (PGE2) and (B) PGD2 on the phagocytosis of fossil fuel-derived particulate matter (PM) <10 μm in aerodynamic diameter (PM10) by human monocyte-derived macrophages. Macrophages were adhered onto coverslips, cultured with either PGE2 10−6 M or PGD2 10−6 M for 10 min, then incubated with PM10 10 μg/mL for 1 h. Phagocytosis of carbon was assessed by image analysis of 50 randomly selected macrophages per coverslip by an operator blinded to exposure and expressed as mean area of carbon (μm2). Preincubation with PGE2, but not with PGD2, suppressed phagocytosis of PM10 (*p<0.05, vs medium control by Wilcoxon matched-pairs signed-rank test). Data are from six separate experiments done at separate times.
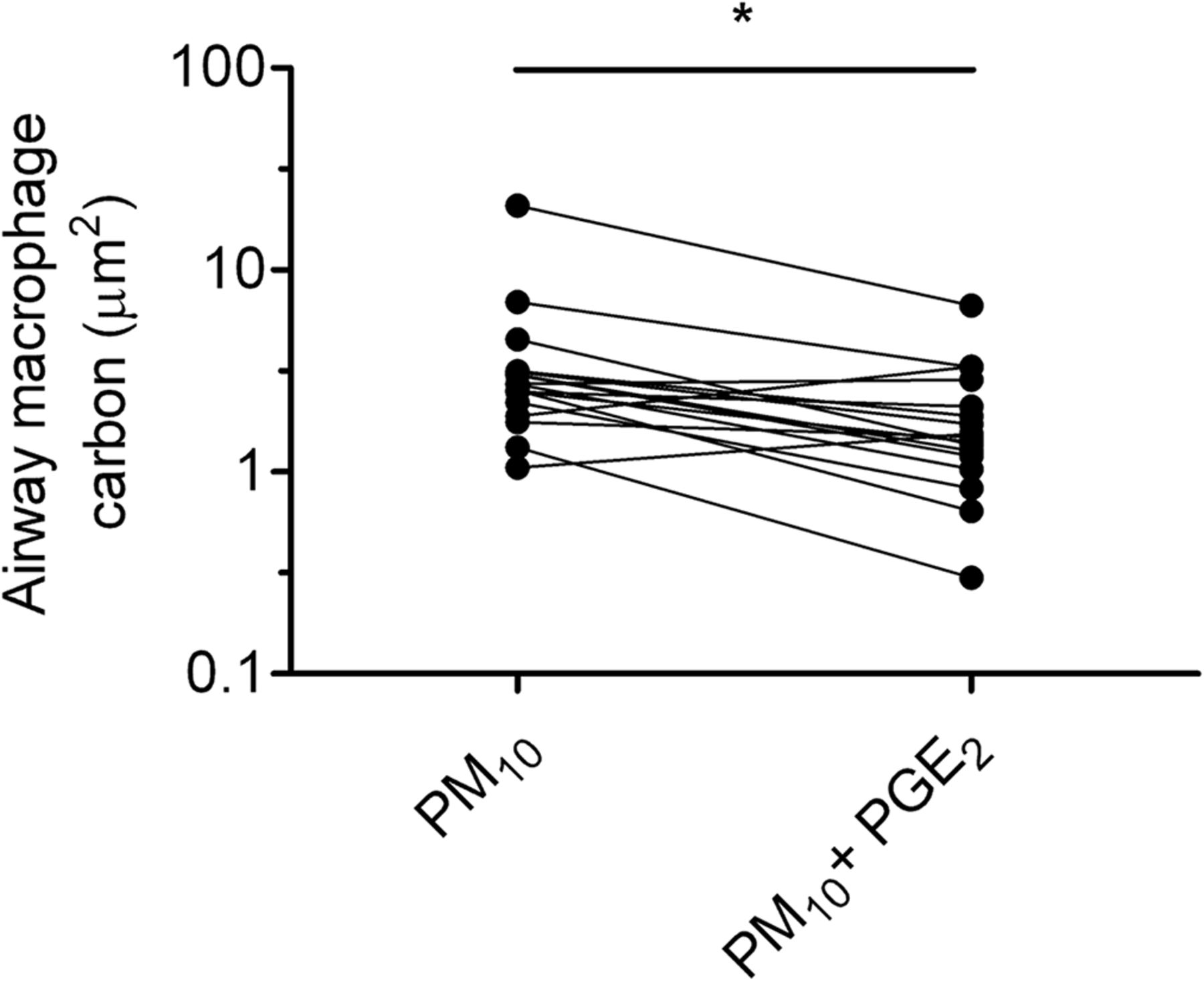
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of prostaglandin E2 (PGE2) on phagocytosis of urban particulate matter <10 µm in aerodynamic diameter (particulate matter10 (PM10)) by rat airway macrophages (AMs). AM were obtained by bronchoalveolar lavage and adhered onto coverslips. Rat AM were cultured with PGE2 10−6 M for 10 min, then incubated with PM10 (10 μg/mL) for 1 h. Phagocytosis of carbon was assessed using image analysis. Paired data are from AM from the same animal. Preincubation with PGE2 10−6 M suppresses PM10 phagocytosis by AM (*p<0.01 by Wilcoxon matched-pairs signed-rank test). Data are from 17 separate experiments in separate animals done at separate times.
Discussion
This study found, compatible with our previous pilot data,14 that AM carbon in children with moderate-to-severe asthma is approximately half that of mild asthmatics and healthy controls. It is unlikely that lower AM carbon in children with severe asthma reflects lower exposure to PM since no difference was found in either modelled exposure to PM at the home address or in the proportion of homes within 50 m of a main road in the subgroup where this was assessed. Children with more severe asthma were older than mild asthmatics, and as expected received higher doses of inhaled corticosteroids. Although there were inverse associations between AM carbon and both age and daily inhaled therapy in asthmatics, these are unlikely to be causal. First, as reported previously in healthy children,14 we found no effect of age on AM carbon in healthy controls in the present study. Second, Fitzpatrick et al1 previously reported that phagocytosis of bacteria by AM in vitro is not attenuated in children receiving regular inhaled corticosteroid therapy for non-asthmatic symptoms. Third, AM phagocytic function is not impaired by β2-adrenoreceptor agonists in vitro.23
Lower AM carbon in children with severe asthma is compatible with previous studies of AM using other phagocytic targets. For example, Huynh et al,3 in a study of adults with severe asthma, reported a reduced number of apoptotic bodies in AM (reflecting decreased phagocytosis of apoptotic cells in vivo24) compared with healthy controls, and in children with poorly controlled asthma, Fitzpatrick et al1 reported an impaired capacity of AM to phagocytose S aureus in vitro. The lack of an effect of mild asthma on the capacity for AM phagocytosis was first suggested by Alexis et al,2 who reported no difference in the capacity of induced sputum AM to phagocytose opsonised yeast in mild asthmatics compared with healthy controls, and second by Lay et al,25 who found that phagocytosis of inhaled particles by AM in vivo was not impaired in mild asthmatics compared with healthy controls. It is unlikely that lower AM carbon in severe asthma is due to less PM inhaled into the lower airway since studies in adults suggest increased lower airway deposition of PM in asthmatics compared with controls.26 ,27 An increased number of AM in asthma could also result in lower AM carbon. To date, AM density in children with asthma has not been studied, but no increase in bronchoalveolar lavage AM concentration has been reported in a murine model of asthma.28
The mechanism causing impaired AM phagocytosis in severe asthma is unclear. Two studies to date have assessed the association between AM phagocytosis in asthma and airway inflammation. Simpson et al29 reported that uptake of apoptotic bronchial epithelial cells by AM in vitro from adults with non-eosinophilic asthma is impaired compared with adults with eosinophilic asthma. By contrast, Alexis et al2 reported that phagocytosis of opsonised yeast particles by AM in vitro is attenuated in adults with eosinophilic asthma compared with a non-eosinophilic asthmatic phenotype. Compatible with Alexis et al,2 we found that AM carbon was indeed lower in children with eosinophilic asthma when defined either by a cut-off of ≥2.5%30 or by a cut-off of >5%2 (p<0.05, see online supplementary figure S2).
PGE2 and PGD2 are putative mediators of impaired phagocytosis in severe asthma since they both are associated with eosinophilic asthma and directly suppress AM phagocytosis in vitro.4 ,31 Profita et al4 reported increased induced sputum PGE2 in asthmatic adults with bronchodilator reversibility >15% and peak flow variability >20%,4 Aggarwal et al5 reported increased induced sputum PGE2 in severe, but not in mild asthmatics, and Fajt et al31reported increased bronchoalveolar lavagefluid PGD2 in severe asthmatic adults. PGE2 in vitro inhibits phagocytosis of opsonised sheep red blood cells and Klebsiella pneumoniae by rat AM,6 and PGE2, and to a lesser extent PGD2, suppresses phagocytosis of apoptotic neutrophils by human monocyte-derived macrophages.8 In the present study, the inverse association between urinary metabolites of PGE2 and PGD2 and AM carbon provides indirect evidence of a role for both mediators in suppressing the phagocytosis of inhaled fossil fuel-derived carbon. Further support for a role of PG is provided by our in vitro studies that show, for the first time, that PGE2 10−6 M markedly suppresses phagocytosis of PM10 by human macrophages and rat AM. The lack of effect of PGD2 at 10−6 M on phagocytosis of PM10 is compatible with Rossi et al,8 who reported that while PGE2 10−6 M suppresses by 60% phagocytosis of apoptotic cells by human monocyte-derived macrophages, PGD2 at a concentration of 10−5 M suppresses phagocytosis by only 20%.8 A limitation of our study is that we did not measure airway PG directly since HPLC-MS is not suitable for small sample volumes obtained using induced sputum, and PGs are rapidly metabolised in the plasma during sampling.32 However, the urinary metabolites measured in the present study are plausible markers of systemic levels since they result from a common metabolic pathway that includes a reduction of the double bond between C-13 and C-14 and oxidation of the hydroxyl group at C-15, producing 13,14-dihydro-15-keto PGs33—metabolites that are excreted unchanged in the urine.22 ,34 Although we have shown that PGE2-mediated suppression of phagocytosis is biologically plausible and have provided indirect evidence for this association in vivo, we cannot exclude a role for ‘intrinsic’ changes in phagocytic capacity of AM in severe asthma. For example, Fitzpatrick et al35 reported that AM from children with severe asthma are characterised by a distinct molecular phenotype that does not have a clear Th1 or Th2 pattern.
In summary, in children with moderate-to-severe asthma with no evidence of lower exposure to fossil fuel-derived PM, we found lower AM carbon compared with mild asthmatics and healthy controls. Indirect evidence was found of increased systemic concentrations of both PGE2 and PGD2—mediators that suppress alveolar macrophage phagocytosis in vitro. Of these, our in vitro studies suggest that PGE2 is a potent suppressor of AM phagocytosis of PM in vivo. The consequences of impaired removal of inhaled PM by AM are unknown in humans, but in mice inhaled particles that are normally non-toxic to wildtype animals are toxic to animals deficient in the receptor required for efficient AM phagocytosis of PM.36
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors REB recruited participants, supervised sampling, analysed sputum samples and performed phagocytosis assays, and wrote the first draft of the manuscript. NM processed sputum samples, supervised phagocytosis assays and contributed to the manuscript. TR recruited participants, supervised sampling and contributed to the manuscript. DHG recruited participants and supervised sampling. ID recruited participants, and supervised sampling and spirometry. EG and LJF provided samples for validation and contributed to the manuscript. LK differentiated monocytes, conducted phagocytosis assays on human cells and contributed to the manuscript. DJL performed geocoding and contributed to the manuscript. MS performed analysis of samples and contributed to the manuscript. HEW recruited participants and supervised sampling and contributed to the manuscript. BB provided pollution exposure estimate calculations and contributed to the manuscript. ISM participated in subject recruitment and sample collection and contributed to the manuscript. FJK and CJG supervised recruitment of participants and sampling. JG devised the study, supervised recruitment of participants, supervised sampling, data analyses, and phagocytosis assays and wrote the first draft of the manuscript with REB.
-
Funding This study was supported by a grant from the Barts and The London Charity. The research was also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval London—Camberwell St Giles Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Additional anonymised data for analysis of carbon in individual macrophages, including stored images, as well as HPLC-MS results from urine and air pollution monitoring data are available upon request to the corresponding author.
Linked Articles
- Airwaves