Article Text

Abstract
Obstructive sleep apnoea (OSA) can result in significant morbidities including the cardiovascular, metabolic and neurocognitive systems. These effects are purportedly mediated via activation of inflammatory cascades and the induction of oxidative stress, ultimately resulting in cellular injury and dysfunction. While great advances have been made in sleep medicine research in the past decades, there are still wide gaps in our knowledge concerning the exact underlying pathophysiological mechanisms of OSA and consequences. Without resolving these issues, the reasons why patients with a similar severity of OSA can have markedly different clinical presentation and end-organ morbidity, that is, phenotype, will continue to remain elusive. This review aims to highlight the recent exciting discoveries in genotype-phenotype interactions, epigenetics, genomics and proteomics related to OSA. Just as PCR revolutionised the field of genetics, the potential power of ‘Omics’ promises to transform the field of sleep medicine, and provide critical insights into the downstream pathological cascades inherent to OSA, thereby enabling personalised diagnosis and management for this highly prevalent sleep disorder.
- Sleep apnoea
Statistics from Altmetric.com
Introduction
Paediatric obstructive sleep apnoea (OSA) is characterised by episodic complete or partial obstruction of the airway during sleep, leading to intermittent oxygen desaturations, recurrent and often sustained rises in carbon dioxide, increases in the magnitude of intrathoracic inspiratory pressures, and in many instances the occurrence of arousals from sleep with resultant sleep fragmentation. OSA is important because it is a common condition affecting 2–5% of the childhood population,1 ,2 and because it can result in significant end-organ morbidities, particularly involving the cardiovascular, metabolic and central nervous systems,3 ultimately resulting in major decrements in quality of life.4–6 Pulmonary hypertension, cor pulmonale, endothelial dysfunction, insulin resistance, dyslipidaemia, neurocognitive impairment, hyperactivity, attention deficits, concentration difficulties, impulsivity, excessive daytime sleepiness, increased healthcare usage, nocturnal enuresis constitute examples of some of the many adverse consequences associated with paediatric OSA that have now been firmly substantiated.7
Pathophysiology of OSA and therapeutic challenges
While adenotonsillar hypertrophy is the most widely recognised attributable contributor to the pathophysiology of paediatric OSA, a combination of anatomical and craniofacial factors (eg, retrognathia and micrognathia, macroglossia or retropositioning of the tongue), lymphoid tissue growth (ie, enlarged tonsils and adenoids), upper airway inflammation and neuromuscular factors are interactively involved, and contribute to the emergence of OSA in children.8 Ultimately, multifactorially induced increases in upper airway collapsibility9 in a child with decreased pharyngeal airway calibre accounts for the vast majority of cases. Accordingly, adenotonsillectomy (AT) is the primary treatment for paediatric OSA for children with adenotonsillar hypertrophy.3 However, while AT is a commonly performed and relatively straightforward procedure, it is still an invasive surgical intervention with attendant intraoperative and postoperative risks. Furthermore, there is a growing realisation that although it is generally an effective treatment leading to major improvements in the severity of OSA in the majority of children, residual significant OSA is still seen in a relatively large subset of children undergoing AT.10 Obesity, presence of severe OSA pre-AT (apnoea hypopnoea index (AHI)>20/hr TST), older age (>7 years), asthma, African ethnicity or a history of prematurity have all been identified as potential risk factors for reduced AT success.10–12 In children who present with minimally enlarged upper airway lymphoid tissues or who manifest residual OSA after AT, positive airway pressure in the form of CPAP or bilevel positive airway pressure (BiPAP) is recommended. Although an effective treatment that has been linked with improvements in cognitive performance,13 adherence can be extremely challenging in children.14 ,15 Non-surgical alternatives or adjuncts to surgery and ventilatory support are critically needed.16 In this context, significant breakthroughs have arisen from translational research: initial observations indicated that proliferative rates of CD3, CD4 and CD8 lymphocytes are increased in mixed cell tonsillar cultures of children with OSA when compared with children with recurrent tonsillitis, that proinflammatory cytokines TNFα, IL-1α and IL-6 are also increased,17 and that neuroimmunomodulatory processes induced by respiratory viruses may be involved.18 These observations raised the possibility that the mechanisms governing lymphoid tissue proliferation in these two conditions, namely OSA and recurrent tonsillitis, may substantially differ, and therefore enable more specific and selective interventions to be developed. In vitro experiments using tonsillar cell cultures showed that when corticosteroids were added to OSA tonsillar cell cultures, decreased proliferative rates and increased apoptosis, as well as reduction in the secretion of IL-6, IL-8 and TNFα emerged.19 Bench to bedside translation of these findings prompted a randomised crossover trial of intranasal budesonide for children with mild OSA, in which 6 weeks of therapy resulted in a reduction in OSA severity, as well as decreases in adenoidal size. Importantly, discontinuation of therapy for 8 weeks did not promote the occurrence of rebound symptoms,20 with similar findings being reported with the use of intranasal fluticasone.21
Tonsils from OSA patients have also been shown to express increased levels of leukotriene receptors 1 and 2 compared with tonsils from children with recurrent tonsillitis.22–24 Although the cellular sources are currently unknown, cysteinyl leukotriene (CysLT) concentrations in the nasal exhaled breath condensate and urine of children with OSA are elevated and correlate with the severity of OSA.25 ,26 The addition of CysLT antagonists to tonsillar mixed cell cultures elicited dose-dependent reductions in cellular proliferation and secretion of the cytokines TNFα, IL-6 and IL-12.27 In an open-label intervention study where children with mild OSA received 16 weeks of montelukast therapy, significant reductions in adenoidal size and respiratory-related sleep disturbances occurred.28 These findings have been confirmed in a recent double-blind placebo-controlled study.29 Use of montelukast together with nasal budesonide for 12 weeks in children who had residual mild residual OSA after AT led to significant improvements in AHI, nadir oxygen saturation and respiratory arousal index, whereas no significant changes occurred over this time period in the control subjects.30 Taken together, these studies emphasise that hypothesis-driven exploration of potential modulators of upper airway lymphadenoid tissue proliferation yield valuable insights, and open new avenues for therapeutic interventions. However, such approaches are cost-ineffective and do not provide a comprehensive map of the orchestrated networks that govern the proliferative properties of lymphadenoid tissues during normal and pathological conditions. Accordingly, implementation of transcriptomic approaches coupled with gene pathway enhancement bioinformatic analyses enabled identification of a discrete set of candidate genes putatively playing a critical role in the pathological chain of events that lead to the emergence of OSA.31 Furthermore, verification of two such putative candidates confirmed the validity of such findings, and further buttressed the possibility of implementing high throughput assessments of therapeutic targets using the in vitro mixed tonsil or adenoid cell culture system coupled with transcriptomic networks-derived candidates.32
Phenotypic variation in paediatric OSA
One of the conundrums that has thus far puzzled researchers and clinicians alike in the field of paediatric sleep medicine, is the question of why at any level of disease severity, there are children who exhibit morbidity, and those who do not. Rather than a clear distinction between disease and non-disease cut-offs, there is now the realisation that the spectrum of OSA is complex, and that disease may occur at the low end of the spectrum, that is, habitual snoring, while absent morbidity may still occur, albeit less likely at the high end of the findings in the polysomnogram (PSG). This has created substantial difficulties among clinicians, particularly since some children who have AHI<1/hrTST on their PSG, and thus, do not fulfil the criteria for significant OSA, but who snore habitually, already display evidence of enuresis or of behavioural, neurocognitive, cardiovascular or metabolic sequelae,33–36 whereas a proportion of children with significantly raised AHI, do not. As with most other diseases, the reasons for this phenotypic variability appear to be multifactorial and environmental, and genetic factors may contribute to the disease phenotype.37
Environmental factors and paediatric OSA
Some examples of environmental factors associated with OSA risk include smoking, pollution, diet and levels of physical activity. Exposure to passive smoking has been shown to be an independent risk factor for habitual snoring in preschool children, and a dose-dependent relationship was identified linking snoring frequency and urinary cotinine concentrations.38 Recently, environmental air quality has also emerged as a significant contributor to the risk of habitual snoring. The frequency of habitual snoring in 6–12-year-old children who resided in neighbourhoods with greatest pollution was three times higher compared to those who resided in neighbourhoods with less pollution. After controlling for confounding factors, the regional variance in the prevalence of habitual snoring was mainly accounted for by an integrated measure of air quality.39
In animal models of OSA, regular non-strenuous physical activity attenuates the spatial learning deficits and oxidative stress induced by chronic intermittent hypoxia (IH) during sleep.40 By contrast, rodents fed a high-fat diet were more susceptible to the cognitive and behavioural deficits induced by IH,41 ,42 that is, nutritional factors can alter recruitment of transcription factors and oxidative-related pathways, and modulate the CNS vulnerability to IH. Interestingly, obese children with OSA are more likely to consume fat-rich diets and less likely to exercise, thereby increasing the magnitude of their cognitive morbidity.43 ,44 Obesity is one of the most significant risk factors for OSA in children and as mentioned previously, the percentage of obese children who have residual OSA following AT is higher than for non-obese children.10–12 Adipose deposits in the neck, thorax and abdomen are thought to result in upper airway narrowing, increased pharyngeal collapsibility and decreased chest wall compliance and functional residual capacity.45 Childhood obesity is becoming increasingly prevalent and is an independent risk factor for adverse cardiovascular, metabolic and neuropsychological consequences. The low-grade inflammatory responses induced by OSA could further interact and potentiate the underlying inflammatory responses due to obesity.46
Conversely, catechin polyphenols, such as found in green tea exert a protective effect in IH-exposed animals, attenuating the IH-induced spatial learning deficits and mitigating IH-induced oxidative stress.47 The inferences from these findings would be that behavioural modifications, such as the encouragement of physical activity and reduction in the dietary intake of foods containing saturated fats and refined carbohydrates may constitute important components of OSA management plans.
Genetic factors and paediatric OSA morbidity
The number of studies examining the associations between specific gene polymorphisms and paediatric OSA-associated morbidity are limited, but have already provided significant inroads into determinants of disease phenotypic variance. For example, several genetic factors can explain, at least in part, some of the discrepancies in functional cognitive performance among children with OSA. Apolipoprotein E (ApoE) functions as a critical regulator of cellular cholesterol deposition and transport, and among its three alleles, the ɛ4 allele is associated with reduced levels of ApoE, thereby resulting in alterations in cellular membrane stability and increased susceptibility to neuronal injury.48 ApoE-null mice and human ApoE-ɛ4 transgenic mice demonstrated increased cognitive deficits when exposed to intermittent hypoxia and sleep fragmentation.49 ,50 As a corollary to such animal studies, the frequency of the ApoE4 allele in children with OSA who manifest deficits in neurocognitive performance was increased when compared with children of similar OSA severity but without evidence of altered cognitive abilities.51 ,52 NADPH oxidase, an enzyme critically involved in free radical formation has been implicated as a potential cause of neuronal cell loss secondary to intermittent hypoxia during sleep.53 The 242 C > T polymorphism in the p22phox subunit of this gene seemingly explains a significant component of the variance in cognitive function phenotype among children with OSA.54
Significant associations between polysomnographic indices and CRP levels have been identified in children with OSA in several studies, even after adjustment for obesity.55–60 Furthermore, the link with neurocognitive morbidity and systemic inflammatory responses was further highlighted by the ability to differentiate children with similar OSA severity using plasma CRP levels into those with cognitive deficits and those without.61 However, there have been a number of subsequent studies which have failed to replicate these results.62–64 To elucidate the potential source of such discrepant findings, a recent genetic association study evaluated CRP and IL-6 gene variants in childhood OSA, and found that certain single nucleotide polymorphisms (SNP) in the CRP and IL-6 genes are related to increased risk for upper airway dysfunction during sleep in US, but not Greek children.65 These latest findings may account for the reported discrepant CRP levels among the two populations as alluded above. The differences in gene variants that significantly associate with OSA between Americans and Greeks might reflect differences in genetic background, environmental exposure or disease phenotypes.
To further evaluate genotype-phenotype interactions, we examined daytime sleepiness as a robust symptom reporter of underlying susceptibility to OSA. By contrast with adults, excessive daytime sleepiness appears not to be as prominent a symptom in children with OSA, with a large proportion of children manifesting ‘sleepiness’ as hyperactive behaviours, impulsivity and impaired attention and concentration on task. Objective measurements of daytime sleepiness using the Multiple Sleep Latency Test have shown that children with OSA have severity-dependent reductions in sleep latency, but that such reductions in latency are highly variable.66 ,67 Furthermore, the magnitude of the reduction in sleep latency is associated with measures of systemic inflammation, such as plasma TNF-α levels.68 While morning TNF-α levels and Epworth Sleepiness Scale scores are increased in the presence of OSA, the substantial variability in these measures can be explained by polymorphisms in the TNF-α gene.69
As mentioned above, OSA is also a risk factor for the occurrence of cardiovascular morbidity. One of the earliest events attesting to such occurrence is the presence of endothelial dysfunction.70 However, the presence of endothelial dysfunction among children with OSA is highly unpredictable.71 Polymorphisms among genes encoding for nitric oxide synthase and endothelin may account for a proportion of such variance in endothelial function.72 ,73 Thus, large-scale multicentre population studies are urgently needed to enable genome-wide exploration of the determinants of end-organ susceptibility in paediatric OSA. Such in-depth analysis of genotype-phenotype interactions in children with OSA may assist in the formulation of categorical risk estimates for each of the morbidities and enable the identification of genetically ‘vulnerable’ patients, that is, precision sleep medicine.
Epigenetics and OSA morbidity
Although it is tempting to explain phenotype variance based on population genetics, it is clear that such approach falls short of accomplishing such goals. The nascent and rapidly evolving field of epigenetics has tremendously expanded our ability to understand phenotypic variation of an ever increasing list of diseases, including cancer, asthma, type II diabetes, cardiovascular disease and autistic spectrum disorders.74 In the last 2 years or so, evidence to suggest that epigenetic changes occur in OSA and may modify specific aspects of OSA-associated morbidity has emerged.
The term ‘epigenetics’ refers to the study of heritable changes in gene expression caused by mechanisms other than changes in DNA sequence. At the molecular level, three main mechanisms have been described: DNA methylation, histone modifications, and non-coding RNAs, including microRNAs (miRNAs), which maintain or change transcription.75 Methylation involves a biochemical modification of the DNA where a methyl group is bound to the carbon-5 position of a cytosine base to form 5-methylcytosine. In mammals, this addition occurs where a cytosine is adjacent to a guanine residue and these locations are referred to as CpGs. Some regions of the genome, especially the promoter regions, are frequently rich in CpGs, and therefore more susceptible to epigenetic regulation. Methylation of CpG sites within these islands plays a crucial role in the transcription and regulation of gene expression, as it can alter transcription factor binding mechanisms, or lead to recruitment of co-repressors.
In an initial effort to examine the hypothesis that changes in DNA methylation may explain the divergent systemic inflammatory responses in children with OSA, Kim et al extracted DNA from peripheral blood mononuclear cells of children with OSA who were matched for gender, ethnicity, BMI and AHI, but differed in their plasma levels of CRP. DNA methylation profiling of a panel of 24 major inflammatory-related genes revealed that children with high CRP levels exhibited increased methylation of the FOXP3 gene compared with children with the same OSA severity with normal CRP levels.76 This was confirmed in a larger second phase case-cohort study that included pyrosequencing of the promoter region of FOXP3. The prevalence of increased FOXP3 methylation was significantly higher in children with OSA and high CRP compared with either children with OSA and normal CRP or control subjects. Furthermore, there was a linear correlation between FOXP3 DNA methylation levels and inflammatory markers, such as CRP and MRP 8/14 levels, as well as with AHI, as a surrogate reporter of OSA severity. FOXP3 is a transcription factor critical to the differentiation of T regulatory lymphocytes (Tregs) which can suppress the activation, proliferation and effector functions of a wide range of immune cells and are important in immune homeostasis. Increased methylation of FOXP3 would reduce the expression of this transcription factor. Indeed, subsequent work revealed that Tregs are less abundant in a subset of children with OSA, and this is associated with altered Th1:Th2 balance towards Th1 predominance.77 Furthermore, percentage of Tregs has been shown to inversely correlate with endothelial function, and a significant negative correlation between Treg function (suppressive ability) and the sleep pressure score, a surrogate measure of sleep fragmentation has also emerged.78 Thus, it is likely that epigenetic modifications of selected candidate genes may contribute to the large dispersion of phenotypes in children with OSA.
As alluded to above, endothelial dysfunction, as assessed using postocclusive hyperaemic responses, is only present in a subset of children with OSA.71 The postocclusive hyperaemic response is dependent on endothelial nitric oxide synthase (eNOS), and DNA methylation has been shown to play an important role in the cell-specific expression of this gene.79 We found that eNOS mRNA expression levels were significantly reduced in the monocytes of children with OSA and with endothelial dysfunction while they remained at control levels among children with OSA but without endothelial dysfunction. Pyrosequencing of the proximal promoter region of the eNOS gene revealed a CpG site located at position -171 that displayed significantly higher methylation levels in the OSA group with endothelial dysfunction. In other words, the presence of abnormal eNOS-dependent vascular responses in children with OSA is associated with epigenetic modifications in the eNOS gene.80
Although this work is clearly preliminary and will require much more comprehensive and extensive evaluations, the epigenetic changes described heretofore add another layer of complexity to the already complex clinical conundrum situation. Indeed, it is possible that conventional treatment may not reverse some of the adverse processes initiated and propagated by OSA, and that novel therapies specifically targeting these epigenetic modifications may be necessary in order to prevent the continued and relentless progression of end-organ injury, and thus prevent premature onset of adult diseases.
Genomic and proteomic approaches to diagnosis
We hope that it is now quite apparent that while PSGs are the current gold standard for the diagnosis of OSA and undoubtedly provide an objective measure of sleep disturbance, measures derived from PSGs are poorly predictive of OSA-associated morbidities. When the cost and investigative burden to the family are also taken into account, it is not surprising that there is considerable incentive to develop alternative tests, and among the available options, there has been great interest in diagnostic biomarkers. A biomarker, as defined by the Biomarkers Definitions Working Group, is ‘a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention’.81 Recent technological advances in fields, such as genomics and proteomics with the concomitant development of sophisticated bioinformatic analytical tools, have resulted in the unbiased exploration of biomarker platforms for diagnostic purposes as the new and rapidly developing paradigm.
The initial genomic studies in the field of sleep medicine primarily involved animal models of OSA. Clear transcriptional effects of intermittent hypoxia and sleep fragmentation were demonstrated in a range of tissues.82 Integrative miRNA-mRNA profiling of adipose tissue in a murine model, revealed that sleep fragmentation induces the activation of several distinct pathways, including those of insulin regulation and diabetes. These results were functionally validated by demonstrating altered glucose and lipid homeostasis in the mice studied.83 Transcriptional profiling of visceral fat from mice exposed to IH revealed the differential expression of over 3000 genes, and gene-gene interaction network analyses showed that the most enriched pathways mapped to metabolic processes, mitochondrion and oxidative stress responses. Once again, the pathophysiological relevance was confirmed by demonstrating that these mice developed dyslipidaemia and underwent significant lipid and protein oxidation within their visceral adipose deposits.84 Exposure to IH will result in the mobilisation of very small embryonic-like pluripotent stem cells from the bone marrow to the peripheral blood.85 In these mobilised cells, a distinct transcriptional response is readily detectable, and gene ontology analysis followed by functional annotation clustering revealed the activation of several organ-specific developmental programmes, including those involved in angiogenesis, central nervous system (CNS) development, and lung development,86 leading to the putative assumption that these cells may play an important role in tissue repair, and thus dampen end-organ damage.
In children, Khalyfa et al performed genome-wide gene expression profiling of leukocytes from the peripheral blood of 20 non-obese OSA patients and compared them with 20 matched controls.31 The altered expression of functionally relevant gene clusters was confirmed and a significant number of these genes mapped to inflammatory processes, providing further support to OSA being a systemic inflammatory disorder. Interestingly, subtle changes in the transcriptome of peripheral blood leukocytes could already be detected in children with primary snoring, compared with matched non-snoring controls, and in these children, altered signalling modules mapping to glucose homeostasis and insulin signalling were detected and confirmed.35 We would surmise that these approaches in exquisitely carefully phenotyped patients may enable identification of candidate pathways underlying unique susceptibility, and offer opportunities for precise categorical risk assessments.
Proteomics
Proteomics is the study of the proteome, namely, the protein complement expressed by a genome87; similar to the transcriptome, characterisation of the changes in protein expression found in the disease condition would be expected to enable the identification of unique pathophysiologically relevant pathways. The complexity of the proteome far exceeds that of the genome due to the vast array of post-transcriptional and post-translational modifications, resulting in a multiplicity of distinct proteins that map to a single gene.
One of the first studies to hypothesise that children with OSA may exhibit different protein expression profiles in their serum was by Shah et al in 2006.88 The proteomic signatures of serum from children with OSA were indeed different from those with primary snoring. Among the multiple candidates, three proteins were identified as capable of diagnosing OSA with 93% sensitivity and 90% specificity. However, the limitations imposed by the existing methodology that was then available precluded identification of the proteins. In adults, plasma levels of malondialdehyde and 8-hydroxydeoxyguanosine have been shown to be significantly higher in OSA patients than in controls, suggesting oxidative stress is greater in OSA patients. Furthermore, levels of these proteins decreased following treatment with CPAP .89 Similarly, a recent study revealed differential serum protein expression in adults with OSA. These proteins were primarily those involved in lipid and vascular metabolic pathways, and the changes varied according to disease severity.90 It is unclear how this line of investigation will evolve and whether exploration of the deep proteome in plasma samples will enable development of a cluster biomarker panel that offers robust and accurate diagnostic capabilities.
In paediatrics, urine is a far more easily obtainable sample compared to blood, so efforts were made to determine whether proteomic differences were similarly present in the urine of children with OSA.
Initial studies used two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) to assess the protein expression in morning urinary samples obtained from children with OSA compared to controls. Mass spectrometry followed by peptide mass fingerprinting analyses identified proteins, namely gelsolin, perlecan, albumin and immunoglobulin, that were differentially regulated, suggesting that OSA may result in alterations in renal glomerular or tubular permeability, or alternatively lead to increased catabolism of these proteins and their subsequent urinary excretion.91 These findings were further expanded using two-dimensional differential in-gel electrophoresis (2D-DIGE) and mass spectrometry-based proteomics followed by validation with western blot analyses or ELISA.92 Of the 16 proteins that were identified as being differentially expressed, some were decreased (eg, Kallikrein 1) in children with OSA, while others were increased (eg, uromodulin). Analysis of uromodulin, urocortin-3, orosomucoid-1 and kallikrein urinary levels showed that a combinational approach that incorporated three or more of these candidate biomarkers yielded a sensitivity of 95% and a specificity of 100%.81 Thus, proteomic approaches have revealed that paediatric OSA is associated with specific and consistent alterations in urinary concentrations of specific protein clusters, and in the future, the signature of a night's sleep could be encapsulated in a cup of urine. To further assess the value of the urinary proteome, nocturnal alterations in urinary neurotransmitters have been explored and identified in paediatric OSA.93 The episodic hypoxaemia and arousals characteristic of OSA likely result in heightened sympathetic activity, and thus will be associated with increased levels of urinary catecholamines such as epinephrine and norepinephrine.94–96 Overnight changes in the levels of three other neurotransmitters, notably increases in GABA, decreases in taurine and β-phenylethylamine (PEA) (rather aptly named for a urinary biomarker) appeared to differentiate children with OSA with neurocognitive deficits from those without such deficits.82 Thus, changes in the patterns of overnight changes among selected urinary compounds could be reflective of mechanisms of neuronal excitotoxicity and dysfunction.
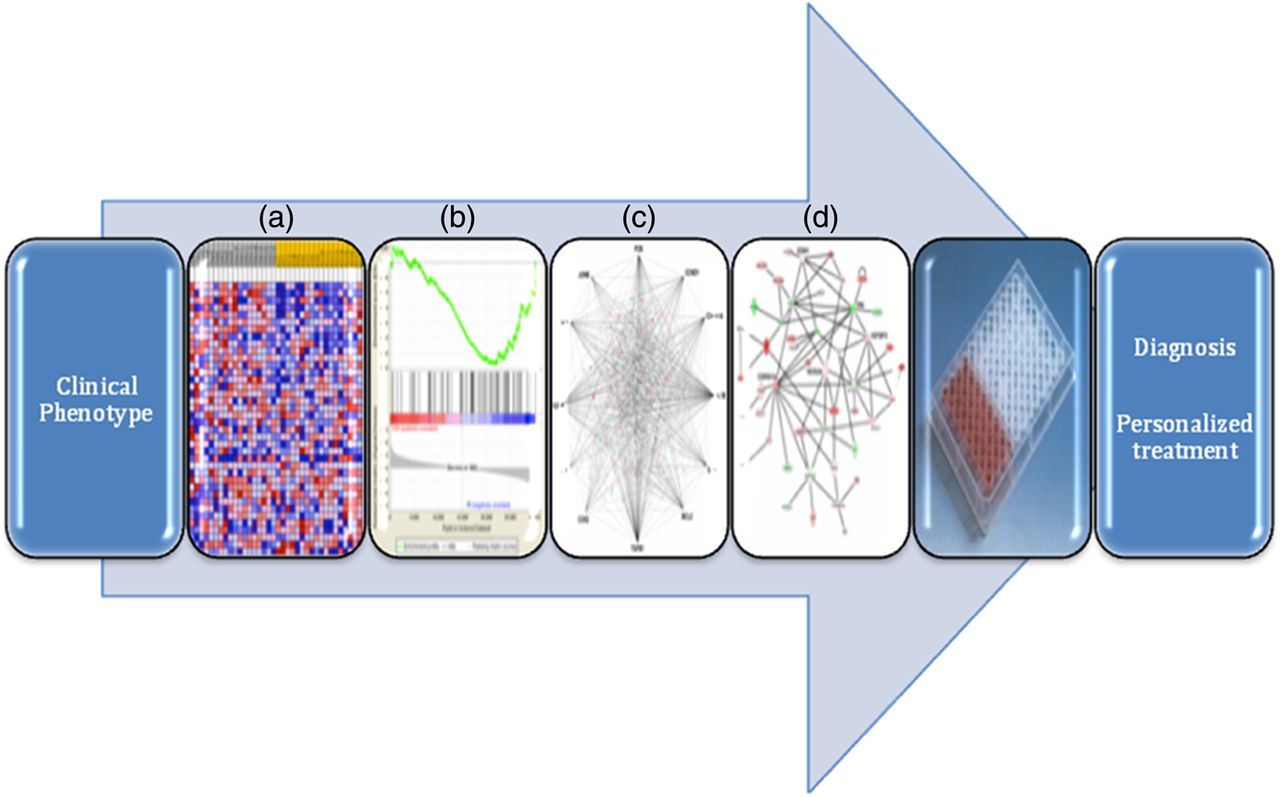
Clearly, the results described above are quite preliminary and need to be validated in larger independent cohorts. However, these findings set a very favourable stage for integration of ‘Omics’ technologies and new bioinformatic approaches in the development of clinically applicable screening or diagnostic strategies for paediatric OSA (figure 1).

{kind=link}
Integration pipeline of phenomics with genomic, proteomic technologies, and bioinformatic approaches in the diagnosis and personalised treatment of OSA. From left to right: (a) transcriptional profiling with the generation of heat maps from microarray analysis, (b) gene set enrichment analysis, (c) integrated network mapping of protein-protein interactions (d) biological network analysis showing nodes (genes) and edges (the biological relationships between the nodes). The intensity of the node colour indicates the degree of upregulation (green) or downregulation (red).
Conclusions
In summary, the substantial progress accumulated over the last decades since the description of OSA in children has enabled an increasingly deeper understanding of the ‘phenomics’ of the disease. Additionally, hypothesis-driven translational studies have resulted in significant breakthroughs in the understanding of the generalisable pathogenetic processes and in the development of novel non-surgical treatments for paediatric OSA. However, such studies have also revealed the unique weaknesses of such methodologies, and their limitations in identifying individual differences and personal disease trajectories. Exponential advances in ‘Omics’ technologies provide us with the ability to interrogate complex biological systems, and should also enable us to derive meaningful results that are immediately and uniquely applicable to each individual patient. Thus, it is not too farfetched to imagine the day when the diagnosis of OSA could be made on a simple urine dipstick, treatment selection become individualised based on a rapid clinic-based susceptibility panel, and delivery of treatment rely on a topical spray of a personalised siRNA cocktail.
References
Footnotes
-
Contributors HLT and LKG drafted the manuscript. DG revised the manuscript, is the guarantor of this work and takes full responsibility for the integrity of data and the accuracy of content. All authors have reviewed and approved the final version of the manuscript.
-
Funding HLT is supported by the NIHR Respiratory Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. LKG and DG are supported by National Institutes of Health grants HL-65270, HL-086662, and HL-107160.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves