Article Text

Abstract
Background Cross-sectional studies implicate neutrophilic inflammation and pulmonary infection as risk factors for early structural lung disease in infants and young children with cystic fibrosis (CF). However, the longitudinal progression in a newborn screened population has not been investigated.
Aim To determine whether early CF structural lung disease persists and progresses over 1 year and to identify factors associated with radiological persistence and progression.
Methods 143 children aged 0.2–6.5 years with CF from a newborn screened population contributed 444 limited slice annual chest CT scans for analysis that were scored for bronchiectasis and air trapping and analysed as paired scans 1 year apart. Logistic and linear regression models, using generalised estimating equations to account for multiple measures, determined associations between persistence and progression over 1 year and age, sex, severe cystic fibrosis transmembrane regulator (CFTR) genotype, pancreatic sufficiency, current respiratory symptoms, and neutrophilic inflammation and infection measured by bronchoalveolar lavage.
Results Once detected, bronchiectasis persisted in 98/133 paired scans (74%) and air trapping in 178/220 (81%). The extent of bronchiectasis increased in 139/227 (63%) of paired scans and air trapping in 121/264 (47%). Radiological progression of bronchiectasis and air trapping was associated with severe CFTR genotype, worsening neutrophilic inflammation and pulmonary infection.
Discussion CT-detected structural lung disease identified in infants and young children with CF persists and progresses over 1 year in most cases, with deteriorating structural lung disease associated with worsening inflammation and pulmonary infection. Early intervention is required to prevent or arrest the progression of structural lung disease in young children with CF.
- Cystic fibrosis
- bronchiectasis
- air trapping
- CT
- bronchoalveolar lavage
- imaging/CT MRI etc
- paediatric lung disease
- asbestos induced lung disease
- asthma epidemiology
- clinical epidemiology
- COPD epidemiology
- interstitial fibrosis
- lung cancer
- mesothelioma
- occupational lung disease
- paediatric asthma
- respiratory measurement
- paediatric physician
- lung physiology
- airway epithelium
- exhaled airway markers
- sleep apnoea
- tobacco and the lung
Statistics from Altmetric.com
- Cystic fibrosis
- bronchiectasis
- air trapping
- CT
- bronchoalveolar lavage
- imaging/CT MRI etc
- paediatric lung disease
- asbestos induced lung disease
- asthma epidemiology
- clinical epidemiology
- COPD epidemiology
- interstitial fibrosis
- lung cancer
- mesothelioma
- occupational lung disease
- paediatric asthma
- respiratory measurement
- paediatric physician
- lung physiology
- airway epithelium
- exhaled airway markers
- sleep apnoea
- tobacco and the lung
Key messages
What is the key question?
Does early cystic fibrosis (CF) structural lung disease detected by CT persist?
What is the bottom line?
CT-detected structural lung disease persists and progresses in the majority of young children with CF and is associated with neutrophilic inflammation and pulmonary infection.
Why read on?
These observations represent the first opportunity to track early CF lung disease using CT. Most children in this study did not have clinically apparent lung disease but had progressive structural lung changes that were associated with pulmonary inflammation and infection. These data support the concept of early intervention to prevent or arrest progressive lung disease.
Introduction
Studies using limited slice high resolution CT (HRCT) scans have demonstrated that infants and young children with cystic fibrosis (CF) diagnosed clinically have dilated airways compared with control infants,1 with bronchiectasis detected in 30% and air trapping in 45%.2 CT-detected changes have been reported in infants without clinically apparent lung disease,3 suggesting that CT-detected structural lung abnormalities may be an early and sensitive marker of the disease status. Following detection by newborn screening, infants with CF at a median age of 3 months have significant lung changes detectable by limited slice HRCT with 19% of infants showing bronchial dilatation and 67% air trapping.4 A cross-sectional analysis of children aged from 2 months to 6 years demonstrated that prevalence and extent of CT-detected structural changes increases during the preschool years.5 Neutrophilic inflammation and pulmonary infection, particularly Pseudomonas aeruginosa, are implicated as contributors to structural lung disease.4 5
Improvement in CT-detected indicators of structural lung disease has been observed following treatment of pulmonary exacerbations in children with CF younger than 4 years of age,6 and children with non-CF bronchiectasis may demonstrate spontaneous improvement of CT abnormalities,7 suggesting that while bronchiectasis is commonly detected in infants and preschool children with CF, a proportion of these children might have reversible airway dilatation. Chest CT has been proposed as an endpoint for clinical trials in young children with CF, therefore longitudinal changes in CT appearance need to be fully understood, particularly changes that occur over the likely duration of early interventional studies.
We hypothesised that early structural lung disease would not be reversible and that persistence and progression of CT-detected abnormalities would be associated with airway inflammation and pulmonary infection. We tested these hypotheses by analysing CT scans acquired 1 year apart in children up to 6 years of age from a newborn screened population and determining associations between persistence and progression of structural changes with simultaneously acquired clinical data including cystic fibrosis transmembrane regulator (CFTR) genotype and symptoms, and bronchoalveolar lavage (BAL)-detected pulmonary infection and inflammation.
Methods
Study population
This retrospective analysis included 143 subjects born between 2000 and 2008, diagnosed with CF following newborn screening and enrolled in the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) early surveillance program (ESP) in Perth and Melbourne, Australia. All aspects of the ESP have been approved by the human research ethics committees at each centre. Parents provided written consent at each assessment. Clinic participation rates for the AREST CF ESP were over 90%, with 85% of study children undergoing annual CT and bronchoscopy with BAL for the duration of the ESP. Pancreatic sufficiency was considered as a single assessment of faecal pancreatic elastase levels at diagnosis, if available, or by requirement for pancreatic enzyme replacement therapy for children transferred to the clinic from other centres. CFTR genotype was classified as ‘severe’ if both mutations were class I, II or III, or ‘mild’ CFTR genotype if at least one mutation was from class IV or V, based on previously published classifications.8–14 The presence of current respiratory symptoms was determined as a dichotomous variable by history and examination by the treating physician on the day of the scan, guided in part by the Kanga score.15
Early surveillance program
The ESP commences soon after diagnosis (median age 3.6 months) with annual CT and bronchoscopy with BAL up to 6 years. The AREST CF ESP was introduced into the Perth clinic in 1999 and into the Melbourne clinic in 2005 and was made available to all children under the age of 6 years in the clinics at that time.
Chest CT
Chest CT scanning was introduced in 2005 and data used in the present study were acquired between 2005 and 2010. The median age at the first ever CT scan was 1.0 year (25th–75th percentile 0.3–2.6 years, range 0.2–5.4 years). Each child in this study had at least two assessments a median of 12 months apart (25th–75th percentile 11–13 months, range 6–18 months) and all available pairs of chest CTs were included in the analysis. Chest CT and BAL were performed as previously described.4 5 Briefly, children were anaesthetised and intubated using a cuffed tube, with a recruitment manoeuvre to reduce procedure related atelectasis.4 5 A volume controlled limited slice scan was performed with three slices taken at end-inspiration (25 cm H2O) and end-expiration (0 cm H2O) as previously described,4 5 with lung volume controlled by the anaesthetist, using a Philips Brilliance 64 (Philips Medical Systems, Eindhoven, The Netherlands) CT scanner in Perth and a Somatron Sensation 16 (Siemens Medical Solutions, Erlangen, Germany) CT scanner in Melbourne. The CT settings used for this limited slice technique are described in the online supplement.
BAL-detected infection
BAL was performed immediately following CT, with three aliquots of 1 ml/kg normal saline instilled into and aspirated from the right middle lobe and one aliquot into the lingula or worst affected lobe identified on CT. The first recovered aliquot from each lobe was sent for microbiological processing to detect bacterial and fungal infections (while viral infections were detected using immunofluorescence or viral culture, they were uncommon (eight infections in nine children) and therefore not included in analyses). BAL samples were considered ‘infected’ if there was a positive culture for at least one predominant organism at any density, ‘mixed oral flora’ if mixed oral flora only were cultured without a predominant organism, or ‘uninfected’ with a negative culture. Details about specific infection with P aeruginosa are given in the online supplement.
BAL-detected inflammatory analyses
The second and third aliquots from the right middle lobe were pooled and processed for inflammatory analyses as previously described,4 5 16 including total and differential cell count, interleukin 8 (IL-8) level measured using ELISA and detection of free neutrophil elastase (NE) activity. The lower limit of detection for IL-8 and NE are 100 pg/ml and 200 ng/ml, respectively. A value half the lower limit of detection was assigned to samples below the limit of detection for statistical analyses.
CT analysis
Images were scored in batches of 5–20 as they became available by a single scorer (CPM) as previously published4 5 on a soft copy reporting station (Agfa Impax TM, Agfa-Gevaert, Mortsel, Belgium) using standard lung window protocols. While the scorer was aware of the diagnosis of CF, scans were assessed without knowledge of the outcome of any previous scans, clinical status, microbiology or inflammatory results. Each scan was divided into six zones (left and right upper, middle and lower zones), corresponding with each axial slice. Each zone was scored for the presence and extent of bronchiectasis and air trapping using a previously reported modification of a CF specific CT scoring system.5 17 Briefly, bronchiectasis was scored on inspiratory images, defined as a bronchus-to-artery ratio >1, or non-tapering bronchus visible in transverse plane. Air trapping was scored on expiratory images, defined as geographical regions of reduced density. Extent in each zone was defined as 0=absent, 1=affecting ≤50% of airways (bronchiectasis) or lung field (air trapping), and 2=affecting >50%. Extent ranged from 0 (absent) to 12 (affecting >50% in all zones). Intra-observer reliability has been previously reported.5 Zones were excluded from analysis if anaesthetic related atelectasis obscured airways in >one-third of the visible segments. If >2 zones were excluded from analysis, the scan was excluded from the study. Demographics and summary data from initial (‘t−1’) and subsequent (‘t’) assessments are presented in table 1.
Demographics of the population and summary data (n=143 children)
Statistical analysis
Each child contributed up to five scans for analysis (49 children contributed two scans, 46 contributed three scans, 32 contributed four scans and 16 contributed five scans). Data were analysed as scan pairs, with bronchiectasis and air trapping during the scan at time t (‘subsequent scan’) as the outcome of interest, in comparison with the scan at t−1 (‘initial scan’), obtained at a median of 1 year prior to time t. Each scan could be represented as both the scan at time t in one pair and as the scan at t−1 in a subsequent pair. Variables that were not normally distributed had a natural logarithmic transformation applied. As neutrophilic inflammatory variables (total cell count, neutrophil count, IL-8 level and NE activity) were highly correlated, they were summarised, guided by a principal component analysis, into a single ‘inflammatory response score’ as the marker of neutrophilic inflammation. Further details are given in the online supplement. Analysis of infection compared ‘infected’ and ‘mixed oral flora’ with ‘uninfected’.
Logistic regression models were used to measure the associations between the persistence of structural lung disease and age, sex, severe CFTR genotype, pancreatic sufficiency, current respiratory symptoms, neutrophilic inflammation and pulmonary infection, assessed where appropriate at both time t and at time t−1. The response variables in the models were the presence of structural lung disease at time t excluding children who did not have bronchiectasis or air trapping present at time t−1. Age at time t−1 was included as a covariate in all analyses.
To examine the associations between the progression of structural lung disease and factors outlined above, linear regression models were used. The data for the extent of structural lung disease were positively skewed and therefore natural logarithmic transformations were applied to the data. The response variables in the models were the log-transformed extent of structural lung disease at time t. The models were adjusted for the extent of structural lung disease at time t−1, age at time t−1 and severe CFTR genotype and excluded children with no detectable bronchiectasis or air trapping at either time t or time t−1.
Further multivariate analyses were performed for both persistence and progression to examine the effects of changes in infection and inflammation status on the presence or change in extent of structural lung disease. The models were adjusted for the extent of structural lung disease at time t−1 (if the outcome was the extent of structural lung disease at time t), age at time t−1, severe CFTR genotype (for progression analyses), infection status at times t−1 and t, and inflammation status at times t−1 and t. The models were chosen to control for the confounding effects of age, severe CFTR genotype (for progression analyses), inflammation and infection.
Repeated measurements on the same child were allowed for in both persistence and progression analyses by using generalised estimating equations. This approach was selected due to the ease of interpretation of results as it focuses on marginal parameters. Similar results were obtained using other models to account for repeated measures. Data were analysed using Stata V.11.0 (Stata Corporation, College Station, Texas, USA). As we did not want the interpretation of our findings to be influenced by the number of tests performed, we have presented all analyses as previously suggested.18
Results
Bronchiectasis
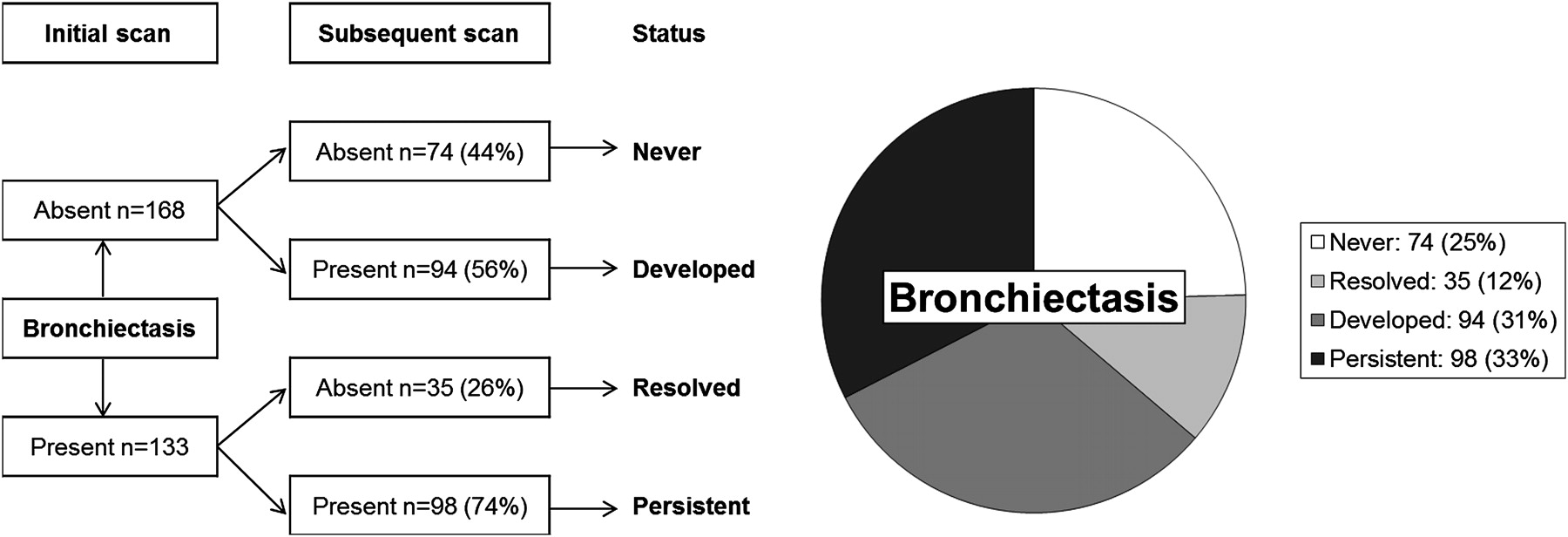
There were 27 (19%) children who never demonstrated bronchiectasis during the surveillance period of this study. Bronchiectasis was never present in 74 pairs (25%), and was detected at either the initial or subsequent scan in 227 pairs (75%) as shown in figure 1. Bronchiectasis was detected at the initial scan in 133 scan pairs. Of these 133 scan pairs, bronchiectasis persisted at the subsequent scan in 98 (74%) (median age 3.1, 25th–75th percentile 1.9–4.2, range 0.2–5.5 years), while 35 (26%) showed apparent resolution of bronchiectasis (median age 2.0, 25th–75th percentile 1.2–3.3, range 0.3–5.4 years). Associations between persistence of bronchiectasis and clinical factors, CT factors, inflammation and infection are shown in table 2.

Bronchiectasis status at initial and subsequent scans.
Associations with persistence of bronchiectasis
Bronchiectasis was detected at either the initial scan or subsequent scan (or both) in 227 paired CT scans. Of these, the extent of bronchiectasis increased from the initial to the subsequent scan in 139 scan pairs (63%), with extent unchanged in 24 scan pairs (11%), and extent decreased in 59 scan pairs (27%). After excluding children who never had detectable bronchiectasis, the median extent of bronchiectasis increased from 1 (25th–75th percentile 0–3) at the initial scan to 2 (25th–75th percentile 1–4) at the subsequent scan. Associations between progression of bronchiectasis and clinical factors, CT factors, inflammation and infection are shown in table 3.
Associations with progression of bronchiectasis
Air trapping
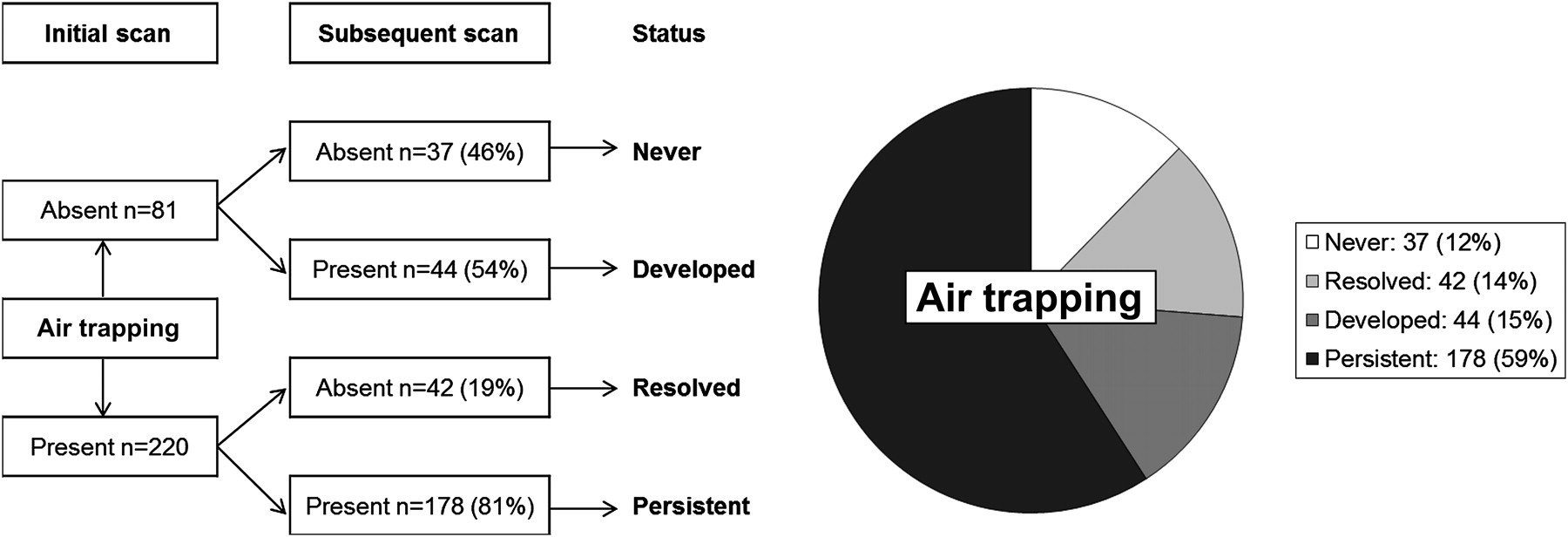
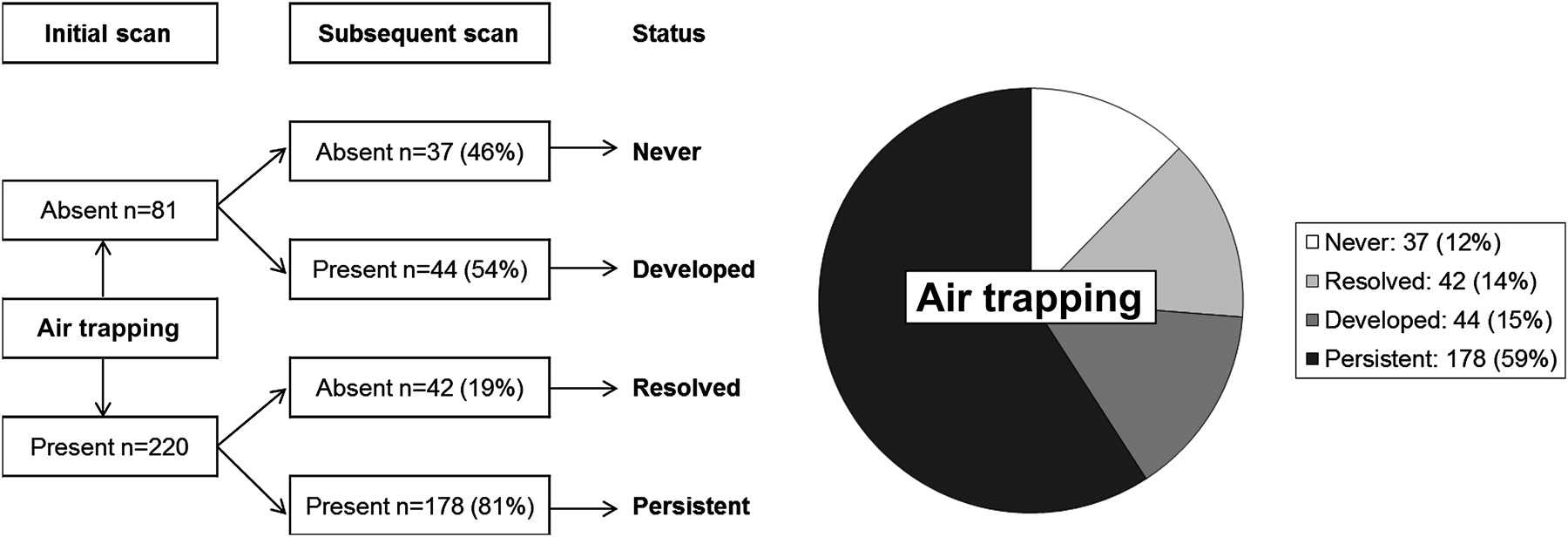
Air trapping was never present in 37 pairs (12%) and was detected at either the initial or subsequent scan in 264 pairs (88%) as shown in figure 2. Air trapping was detected at the initial scan in 220 scan pairs. Of these 220 scan pairs, air trapping persisted at the subsequent scan in 178 pairs (81%) (median age 1.97, 25th–75th percentile 0.97–3.17, range 0.2–5.5 years), while 42 pairs (19%) (median age 2.24, 25th–75th percentile 1.24–3.44, range 0.3–5.4 years) showed apparent resolution of air trapping. Associations between persistence of air trapping and clinical factors, CT factors, inflammation and infection are shown in table 4.
{kind=link}
{kind=link}
Air trapping status at initial and subsequent scans.
Associations with persistence of air trapping
Air trapping was detected at either the initial scan or subsequent scan (or both) in 264 (88%) paired CT scans. Of these, the extent of air trapping increased from the initial to the subsequent scan in 121 scan pairs (46%), with extent unchanged in 36 scan pairs (14%), and extent decreased in 107 scan pairs (41%). After excluding children who never had detectable air trapping, the median extent of air trapping at both the initial scan (25th–75th percentile 0-5) and the subsequent scan (25th–75th percentile 0–6) was 3. Associations between progression of air trapping and clinical factors, CT factors, inflammation and infection are shown in table 5.
Associations with progression of air trapping
Discussion
This is the first longitudinal assessment of CT-detected structural lung disease in young children with CF detected by newborn screening. The observations provide a unique insight to the early development of CF lung disease and factors associated with the persistence and progression of structural changes. We found that neutrophilic inflammation and pulmonary infection were independently associated with progression of bronchiectasis, with neutrophilic inflammation associated with progression of air trapping. Importantly, neutrophilic inflammation at the initial scan was associated with subsequent progression of structural lung disease and might therefore be an early predictor of significant future lung disease. These findings are in agreement with earlier cross-sectional studies that implicated neutrophilic inflammation and pulmonary infection as risk factors for structural lung disease.4 5 We also identified an association between air trapping at the initial scan and subsequent persistence and progression of bronchiectasis, which may offer some insights into the pathophysiology and natural history of different structural lung disease components. Further analyses are required to identify whether early air trapping can predict those children who will later develop bronchiectasis at an early age.
In addition to associations with neutrophilic inflammation and pulmonary infection, we identified an independent association between severe CFTR genotype and progression of bronchiectasis and air trapping. Previous studies have identified associations between mild CFTR genotypes and lower rates of infection with P aeruginosa and other pathogens,14 19 and chest radiography and lung function decline is faster in patients with severe CFTR mutations20 with poorer lung function seen in those with the most severe mutations.21 However, close correlations between clinical pulmonary phenotypes and CFTR genotype have been elusive.13 To our knowledge, no other studies have examined the link between CFTR genotypes and structural lung disease so early in life in a newborn screened population. The causal pathways that link CFTR dysfunction with structural lung disease are poorly understood, but studies such as ours provide information regarding the earliest pathobiological events in the lung, and determining relations with structural changes are likely to be a key to defining these pathways. Further studies in larger populations are required to confirm this association.
There are some limitations to this study. To determine whether structural lung disease was common in infancy we initially chose a three-slice HRCT method as a screening tool using a low dose protocol, before more extensive imaging techniques with low radiation exposures had been adequately validated. In practice, our estimates of persistence and progression of bronchiectasis are likely to have been underestimated because limited slice scans underestimate this element of structural change,22 and this limitation is likely to have resulted in misclassification of apparently resolved bronchiectasis. Thus, using this radiological method, we cannot say with certainty whether early bronchiectasis truly resolved in any infants or preschool children, though it is interesting to note that of the 35 pairs that showed apparent resolution of bronchiectasis, 13 had further annual scan(s) and of these, 12 children (92%) demonstrated recurrence of bronchiectasis. Despite this, we demonstrated that children with apparent resolution of bronchiectasis were less likely to have worsening neutrophilic inflammation and infection at the subsequent scan. Scans were assessed by an experienced paediatric thoracic radiologist (CPM) as previously described.4 5 While we were unable to make the size and resolution of each scan identical and it was not possible to blind the scorer to the approximate age of the child, we do not believe that an age bias is likely because at the time of the assessment the scorer was unaware of whether the images represented the initial or subsequent scan of any given pair from children in a relatively wide age range. While the CT scans were available to treating clinicians and evidence of lung disease might lead to more aggressive clinical management, this would be expected to reduce the likelihood of demonstrating persistence and progression as we have done so in this study.
The results of this study have implications for the clinical management of young children with CF. We observed that neutrophilic inflammation at the initial scan was associated with subsequent progression of both bronchiectasis and air trapping. This observation suggests a possible role for anti-inflammatory therapies early in life. However, the effectiveness of anti-inflammatory agents for preventing, delaying or reversing structural lung disease in young children with CF is unknown and needs to be investigated in appropriately designed, prospective early intervention trials. Importantly, respiratory symptoms at the initial scan were not associated with increased rates of persistence or progression of bronchiectasis or air trapping, and therefore symptoms cannot be relied upon to identify children at risk of further deterioration in structural lung disease. While respiratory symptoms at the subsequent scan were associated with persistence and progression of bronchiectasis, the overall prevalence of symptoms was low in this cohort and most children with persistent or progressive disease did not have respiratory symptoms at the time of their subsequent scan. Associations between pulmonary infection at the second scan of a pair and structural lung disease emphasise the importance of early detection of infection since infection is an important determinant of inflammation23 and eradication, particularly of P aeruginosa, is usually successful when detected early.24
This study contributes to our understanding of CT-detected structural lung disease in young children and has implications for clinical trials in young children with CF. CT as an outcome measure for clinical trials for infants and pre-school children with CF shows great promise, particularly since traditional outcome measures used by regulatory agencies are not appropriate for young children.25 The findings from this study suggest a low rate of spontaneous resolution of structural changes with evidence of disease progression over 1 year. Therefore, CT could provide direct measurements of CF lung disease for use in short term (1–3 years) intervention studies of potentially disease modifying therapies in infants and preschool children, groups currently excluded from early intervention studies due to the absence of acceptable trial endpoints. Further research using CT as an outcome measure should focus on the use of ultra-low dose volumetric CT scans to improve our understanding of lung disease in CF and to develop strategies to optimise image quality, minimise radiation dose and determine the best method to quantify the extent of early CT detected structural lung disease in young children. While we acknowledge the small inherent risk associated with exposure to ionising radiation using chest CT, the limited slice approach used in this paper involves a very low radiation exposure of approximately 0.28–0.40 mSv as previously reported.4 5 Using the latest scanning techniques and software we are now able to obtain high quality low dose full inspiratory and expiratory volumetric scans that provide more complete information about structural lung disease, and research to determine the best methods for quantification of structural lung disease from these scans is ongoing.
In conclusion, we have shown that early structural lung disease, measured by limited slice chest CT, persists in most children once detected, with progression of disease associated with worsening neutrophilic inflammation, pulmonary infection and severe CFTR genotype. Since lung disease can commence very early in life, and early inflammation is associated with subsequent disease progression, interventions to prevent structural lung disease or arrest progression must be introduced soon after diagnosis.
Acknowledgments
The AREST CF would like to thank the contributions of Princess Margaret Hospital for Children, The Telethon Institute for Child Health Research, Royal Children's Hospital Melbourne and Murdoch Children's Research Institute. We would also like to thank the participants and their families who contributed to the AREST CF programme. We also acknowledge the support of the Cystic Fibrosis Foundation, Australia. The full authorship includes the following members of the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF): Luke Berry, Rosemary Carzino, Barry Clements, Luke Garratt, Graham Hall, Jo Harrison, John Massie, Srinivas Poreddy, Shannon Simpson, Billy Skoric. The full membership of AREST CF is available at http://www.arestcf.org. We acknowledge Dr Marcel van Straten from the Erasmus MC, Rotterdam, The Netherlands and Zoe Brady from The Alfred Hospital, Melbourne, Australia, for assistance with radiation dose calculations for our CT scans.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
See Editorial, p 471
Funding AREST CF is funded by the Cystic Fibrosis Foundation Therapeutics, Inc (Sly040A, Stick090A), USA; the Australian Cystic Fibrosis Research Trust; and the National Health and Medical Research Council (513730), Australia. LSM is funded by a PhD scholarship from the National Health and Medical Research Council and supplementary scholarships from the University of Western Australia and the Australian Cystic Fibrosis Research Trust. This research was presented in part at the European Cystic Fibrosis Society Conference, European Respiratory Society Annual Scientific Meeting and Thoracic Society of Australia and New Zealand Annual Scientific Meeting, supported by the Lung Institute of Western Australia, the University of Western Australia and the Thoracic Society of Australia and New Zealand.
Competing interests None.
Ethics approval The study was approved by the ethics committees of the Princess Margaret Hospital, Perth, and Royal Children's Hospital, Melbourne.
Provenance and peer review Not commissioned; externally peer reviewed.