Article Text

Abstract
Chronic obstructive pulmonary disease (COPD) is characterised by an inflammatory response by the lungs to inhaled substances such as cigarette smoking and air pollutants. In addition to the pulmonary features of COPD, several systemic effects have been recognised even after controlling for common aetiological factors such as smoking or steroid use. These include skeletal muscle dysfunction, cardiovascular disease, osteoporosis and diabetes. Individuals with COPD have significantly raised levels of several circulating inflammatory markers indicating the presence of systemic inflammation. This raises the issue of cause and effect. The role of tumour necrosis factor α in COPD is thought to be central to both lung and systemic inflammation and has been implicated in skeletal muscle dysfunction, osteoporosis and type 2 diabetes. It has been hypothesised that inflammation in the lung results in ‘overspill’ into the circulation causing systemic inflammation. There is supportive evidence that protein movement can occur from the lung surface to the systemic circulation. Evidence from inhaled substances such as air pollutants and cigarette smoke has demonstrated a temporal link between the inflammatory process in the lung and systemic inflammation. Also, studies have shown alterations in circulating inflammatory cells in patients with COPD compared with controls which may reflect the effects of inflammatory mediators (derived from the lung) on circulating cells or the bone marrow. This paper considers the concept of ‘overspill’ in depth, reviews the current evidence and highlights problems in generating direct evidence to support or refute this concept.
- COPD
- systemic inflammation
- comorbidity
- overspill
- cytokines
- systemic disease and lungs
Statistics from Altmetric.com
Introduction
Chronic obstructive pulmonary disease (COPD) is characterised by an intense inflammatory process in the airways, parenchyma and pulmonary vasculature. The lung inflammatory response is characterised by increased numbers of neutrophils,1 macrophages and T lymphocytes2; augmented concentrations of proinflammatory cytokines such as leukotriene (LT)B4,3 interleukins (IL)-1, 6 and 84 and tumour necrosis factor α (TNFα)5; and evidence of oxidative stress. It has been recognised that systemic features and other diseases are more common in COPD,6 including skeletal muscle dysfunction,7 cardiovascular disease,8 osteoporosis9 and diabetes,10 all of which are thought to have a similar inflammation-based pathophysiology to COPD. Indeed, COPD is associated with evidence of systemic oxidative stress,11 activation of circulating inflammatory cells12 and increased plasma levels of proinflammatory cytokines.13 A systematic review supported this and concluded that individuals with COPD had significantly raised levels of several markers of inflammation including C-reactive protein (CRP), IL-6, fibrinogen, activated leucocytes and TNFα, confirming the presence of systemic inflammation.14 However, whether this represented independent physiological processes, common generalised processes or ‘overspill’ inflammation from one organ to another remains uncertain. This review examines the evidence and specifically addresses the feasibility of ‘overspill’ and its complexity.
Role of systemic inflammation in systemic features of COPD
The role of TNFα in COPD is thought to be central to both lung and systemic inflammation.15 16 Plasma TNFα and its soluble receptor are increased in patients with COPD.17 18 Di Francia et al16 showed that, when serum TNFα was measured by immunoradiometric assay, there were significantly higher (p<0.001) levels in patients with COPD with weight loss than in healthy controls. Similarly, Karadag et al19 reported higher serum TNFα levels in both stable COPD patients and those with an exacerbation compared with controls, and serum TNFα levels correlated with disease severity.20 Furthermore, circulating monocytes and bronchoalveolar lavage T lymphocytes harvested from patients with COPD produce more TNFα than those from healthy controls.21 22 The role of TNFα in muscle wasting is controversial, with some studies showing a relationship23 while others do not.24 25 However, measurements of TNFα do not necessarily equate to function as rapid receptor binding will remove it from the biological fluid. TNFα may affect muscle cells in a number of ways. In differentiated myocytes studied in vitro, TNFα activates the transcription factor nuclear factor-κB and degrades myosin heavy chains through the ubiquitin/proteasome complex.26 Studies have shown that dysregulation of the ubiquitin/proteasome system contributes to the loss of muscular mass caused by sepsis or tumours in rats,27 suggesting that this is a likely mechanism in COPD. In addition, TNFα can induce apoptosis in several cell systems including skeletal muscle in patients with COPD with weight loss.6 Alternatively, the effects of TNFα may be less direct inducing other proinflammatory cytokines that are likely to contribute to the persistence and amplification of the inflammatory cascade.26 Patients with COPD with increased resting energy expenditure and reduced fat-free mass also have increased levels of acute phase reactant proteins and other inflammatory cytokines in their serum such as CRP, lipopolysaccharide binding protein and IL-8,13 indicating a more general inflammatory response.
Bolton et al9 confirmed that osteopenia was a feature of COPD and also associated with an increase in circulating TNFα. Low bone mineral density and high fracture rates have been demonstrated in patients with COPD not receiving systemic glucocorticoids28 and impaired forced expiratory volume in 1 s (FEV1) has been shown to be an independent predictor of osteoporosis in studies of patients with and without COPD.29 Furthermore, TNFα and IL-1 have been implicated in the pathophysiology of osteoporosis,30 providing a putative direct link.
Despite this evidence and hypothesis, treatment with TNFα inhibitors has not shown significant short-term benefit in patients with COPD,31 although etanercept may reduce hospital admissions.32 This may suggest that TNFα has little role in COPD. However, COPD is a highly complex inflammatory disease in which many other cytokines and mediators are involved, and blocking a single cytokine does not necessarily lead to a clinically significant effect.33 Alternatively, the relatively low levels seen in COPD and the slow progression of the disease may mean that TNFα inhibition requires a long period before any effect (other than on an acute event) can be clearly identified or dismissed.
Persistent systemic inflammation has also been linked to atherosclerosis, ischaemic heart disease, stroke and cardiovascular mortality. Circulating CRP is an independent predictor of vascular death34 and rises during an acute exacerbation of COPD. Reduced FEV1 is a marker of cardiovascular mortality independent of age, gender and smoking history35 and is also related to plasma CRP levels,36 again suggesting a pathophysiological link. Patients with COPD have elevated plasma fibrinogen levels37 which are associated with an increased prothrombotic risk. Exacerbations are associated with a rise in serum IL-6 levels leading to a further rise in plasma fibrinogen and increasing CRP production. Thus, acute infection leads to changes that may have a role in predisposing to coronary heart disease or stroke and hence explain the increased mortality observed within a few months of hospital admission.38
These data indicate clearly that common cytokines or pathways are involved in the pathophysiology of COPD and its comorbidities, although the exact mechanisms are far from clear and direct evidence of ‘overspill’ is lacking.
Protein movements between the lung and circulation
Evidence of protein movement from serum to lung secretions
There is abundant evidence that protein movement can take place from the systemic circulation to lung secretions. Gorin et al39 measured the flux of albumin between the vascular space and the pulmonary interstitial and luminal lining fluids in 20 adult sheep with chronic lung lymph fistulas. They concluded that proteins present in alveolar lavage fluid and also present in plasma reach the alveolar space by a simple diffusion process which was relatively free into the interstitium although markedly restricted by the epithelial membrane. Radiolabelled albumin injected into the peripheral circulation subsequently appears in lung secretions.40 Also, a rise in the concentration of α1-antitrypsin in lung secretions occurs following intravenous administration of the protein to deficient subjects.41 42 The secretion to serum concentration ratio of proteins is dependent on the plasma concentration, is inversely proportional to protein size indicating a degree of filtration and is influenced by the presence of inflammation which increases epithelial leakability.43
Evidence for movement from the lungs to the systemic circulation
Direct evidence
There is evidence that proteins can move in the opposite direction from the lung surface to the systemic circulation. Smith et al44 administered aerosols of plasma-derived α1-antitrypsin to the lungs of dogs and sheep. The protein was subsequently found in the plasma of dogs, rose slowly to a maximum value at 48 h, remained raised from 48–72 h and then disappeared slowly by 144 h. The aerosolised α1-antitrypsin was also found in the lymph and plasma of sheep with a reverse gradient supporting retrograde movement. Vogelmeier et al45 showed that both recombinant secretory leucoprotease inhibitor and recombinant α1-antitrypsin appeared in lung lymph of sheep following aerosolisation. Finally, Hubbard et al46 administered aerosolised human plasma α1-antitrypsin to 12 patients with homozygous Z type α1-antitrypsin deficiency and mild to moderate emphysema and to anaesthetised sheep with indwelling thoracic lymph duct catheters for direct assessment of lung permeability. Aerosolised α1-antitrypsin diffused across the respiratory epithelium and entered lung interstitial lymph (in sheep) and was detected in the systemic circulation (in both sheep and humans).
In addition, aerosolised insulin has a rapid systemic therapeutic effect.47 These data clearly confirm the ability of proteins in the airway to move into the systemic circulation, although this is also likely to be influenced (at least in part) by factors outlined above that determine protein movement in the opposite direction.
Evidence that inhaled substances can influence systemic inflammation
In chronic diseases the relationship between local and systemic inflammation is in a stable state. However, the temporal relationship between factors that change local and systemic inflammation provides clearer evidence of an ‘overspill’ phenomenon.
For instance, there is evidence that inhaled substances such as air pollutants can lead to a subsequent systemic inflammatory response. Alveolar macrophages (AM) are the most likely link between the inflammatory process in the lung and the systemic response because they are cells responsible for ingesting and clearing inhaled particles.48 Human and animal studies have shown that phagocytosis of these particles by AM leads to pulmonary inflammation with an increased number of activated AM.49 The interaction of AM with atmospheric particles increases their phagocytic activity, oxidant production and the release of inflammatory mediators such as TNFα.50 51 This is accompanied by raised levels of circulating cytokines, systemic inflammation and microvascular endothelial dysfunction in the systemic circulation.52–55 It has been suggested that, following phagocytosis of particulate matter, cytokines released by activated AM act on the bone marrow to mobilise platelets and leucocytes. Indeed, studies have shown that particle exposure leads to an acute leucocytosis in humans56 and animals,57 supporting this concept. The instillation of mediators secreted by AM exposed to particulate matter with diameter <10 μm (PM10) ex vivo into lungs produces a bone marrow response similar to that produced by instilling the particles themselves into the lungs.57 58 This suggests that AM are capable of initiating both a local and systemic inflammatory response when PM10 are deposited in the lung. In support of this, Van Eeden et al55 found that circulating levels of IL-1, IL-6 and granulocyte macrophage colony stimulating factor (GM-CSF) were raised in subjects exposed to high levels of PM10 during an episode of acute air pollution. The results showed that a range of different particles stimulate AM to produce proinflammatory cytokines, and these cytokines are also increased in the blood of subjects during an episode of acute atmospheric air pollution indicating a temporal link between these cytokines and the systemic response.
Salvi et al59 exposed human healthy volunteers to air and diluted diesel exhaust for 1 h. Blood sampling and bronchoscopy were performed 6 h after each exposure to obtain airway lavages and endobronchial biopsies. There was a significant increase in neutrophils and B lymphocytes in the bronchial lavage following exposure to diesel exhaust, together with increases in histamine and fibronectin. The bronchial biopsies obtained 6 h after exposure to diesel exhaust showed a significant increase in neutrophils, mast cells, CD4+ and CD8+ T lymphocytes together with upregulation of the endothelial adhesion molecules intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. At the same time, significant increases in neutrophils and platelets were observed in peripheral blood following exposure to diesel exhaust, suggesting stimulation of marrow release. These data provide clear evidence that inhaled pollutants can lead to a temporal association between the expected pulmonary response and an identified systemic inflammatory effect.
Cigarette smoking is the most important cause of COPD, although the fact that only a proportion of smokers develop clinically important airflow obstruction suggests a genetic predisposition. Rusznak et al60 examined cultured human bronchial epithelial cells exposed to cigarette smoke from never-smokers, and smokers with and without COPD. They concluded that bronchial epithelial cells from smokers with COPD were different, showing greater response to the effects of cigarette smoke by increasing transepithelial permeability and reducing the protective role of cellular glutathione (an important intracellular antioxidant). Also, there was increased release of key proinflammatory mediators (IL-1β and soluble intercellular adhesion molecule-1) from the COPD cells in response to the smoke challenge. There have been conflicting data on the response of AM to cigarette smoke. Some authors have reported reduced cytokine production following exposure to cigarette smoke.61 However, Terashima et al62 demonstrated that AM, when stimulated by cigarette smoke, produce factors such as TNFα, IL-1, IL-6 and IL-8, as well as haematopoietic growth factors such as GM-CSF and G-CSF, which are capable of stimulating the proliferation and release of polymorphonuclear leucocytes and monocytes from the bone marrow and hence may account for the blood neutrophilia of smokers.63
Overall, these experimental data indicate that AM and bronchial epithelial cells are critically important in processing inhaled noxious gases and particles. Many of the mediators they produce and/or their effects are also identified systemically in COPD, suggesting that these mediators have been generated in and released from the lung into the circulation contributing to the systemic inflammatory response. Although other cells are able to produce these mediators, the importance of AM in producing systemic inflammation has been demonstrated best using animal models. AM-depleted rats show a substantially reduced systemic inflammatory response following acute alveolar hypoxia compared with controls,64 indicating a key role for this cell.
Experimental data also exist demonstrating a clear relationship between the systemic inflammation induced by instillation of air pollution particles in the lung and progression of atherosclerosis. Suwa et al65 showed that, when Watanabe Heritable Hyperlipidaemic rabbits (that develop atherosclerosis naturally) were exposed to ambient particles, they demonstrated a brisk systemic inflammatory response which was associated with progression of their atherosclerosis. Furthermore, the extent of the atherosclerotic burden was directly proportional to the number of AM that contained the particulate matter. These data again suggest a direct link between lung and systemic inflammation and subsequently inflammation-related comorbidity.
Alternatively, there is evidence that inhaled ultrafine particles are able to translocate from the lung into the systemic circulation in humans and animals and directly activate a systemic response.66 In animal studies, intratracheal instillation of diesel exhaust particles promoted femoral venous thrombosis in a dose-dependent manner. Furthermore, the direct addition of diesel exhaust particles to untreated blood also caused platelet aggregation. These prothrombotic effects persisted for up to 24 h after instillation and provide a plausible mechanistic explanation for the epidemiologically established link between air pollution and acute cardiovascular effects.
Indirect evidence
The lung inflammatory response is characterised by increased numbers of neutrophils, macrophages and T lymphocytes and augmented concentrations of proinflammatory cytokines, which are similar to changes found in the systemic circulation. Studies have shown alterations in circulating inflammatory cells in COPD which may reflect the effects of inflammatory mediators derived from the lung on circulating cells or the bone marrow. For instance, Burnett et al67 showed that neutrophils isolated from patients with COPD had enhanced chemotaxis in response to a chemotactic peptide (suggesting that they had been primed) and increased spontaneous extracellular proteolysis. Noguera et al68 found that patients with COPD showed abnormal expression of neutrophil and endothelial adhesion molecules, and reported downregulation of a G-protein subunit involved in the intracellular transduction pathway. The same authors12 found that the production of reactive oxygen species was higher in circulating neutrophils harvested from patients with COPD than from smokers with normal lung function or non-smokers. These studies of enhanced neutrophil function in COPD also support a systemic influence related in some way to the lung abnormality.
Blood lymphocytes isolated from patients with COPD have been less well studied, although abnormal function of circulating lymphocytes in COPD has been reported69 which relates significantly to disease severity.
Monocytes also accumulate in the lungs of smokers in response to cigarette smoke.70 It is likely that smoke-activated macrophages release monocytic chemokines (eg, monocyte chemoattractant protein-1) into the peripheral blood.71 Besides being chemoattractant, these chemokines can also contribute to the subsequent priming of—and increased expression of—CD43 and CD11b receptors on circulating monocytes.72 Activated monocytes have also been implicated in the development of atherosclerosis73 which is increased in patients with COPD.
Patients with COPD have higher circulating levels of IL-6 than controls.74 This cytokine is a potent stimulator of CRP production by the liver and may account for the increase in circulating CRP found in patients with COPD.75 In stable COPD, plasma concentrations of CRP are related to mortality in patients with mild to moderate disease76 but not in those with severe and very severe disease.77 Raised CRP relates to health status, exercise capacity and body mass index (BMI)78 as well as cardiovascular disease,34 indicating a clear association with both lung-related and systemic features.
Surfactant protein D (SPD) is a large multimeric collagenous glycoprotein produced mainly by type 2 pneumocytes in the lungs, although endothelial cells and glandular cells in the gastrointestinal tract can also produce small amounts.79 In a pilot study,80 serum SPD concentrations were found to be increased in patients with COPD and related to disease severity and symptoms. Since the lung is the major source of SPD production, this observation suggests that, with lung injury, SPD and (by implication) other locally produced proteins may leak from the lung compartments into the systemic circulation.
Clinical evidence
Sapey et al81 found that sputum levels of IL-1β, TNFα and LTB4 correlated negatively with BMI, supporting the hypothesis that an increase in lung inflammation is associated with a lower body weight and previous studies have linked low BMI with systemic inflammation.23 This is an important observation, especially as TNFα has been implicated in the muscle loss of COPD.13 16 21 However, the lung TNFα concentration did not correlate with the plasma level. The absence of a clear relationship between lung and systemic TNFα would be taken as lack of direct evidence of ‘overspill’, thereby questioning this as a mechanism.
Difficulties in proving the concept of ‘overspill’
Clearly, protein movement can occur from the airway into the circulation and circumstantial evidence exists indicating that airways disease and inflammation are associated with inflammatory changes in the circulation and an increase in inflammation-related comorbidity. It is therefore understandable that the concept of inflammatory cytokine ‘overspill’ should be implicated. However, a lack of correlation between airway cytokine concentrations and those in the circulation has failed to support this concept,82 83 although studies to date have involved small numbers of patients and may therefore have lacked sufficient power to support the ‘overspill’ theory.
Nevertheless, this lack of correlation does not mean that ‘overspill’ has not occurred as many factors will influence the results. First, a protein in the airway has to pass into the interstitium, and hence lymph, in order to reach the circulation. This process will be dependent upon the ‘leakability’ of the epithelium and protein size (as shown for the reverse process from plasma to the airway). Second, facilitated transport may occur by processes including epithelial basolateral secretion of the same protein84 or migration of activated macrophages from the airway into the interstitium or vice versa, thereby secreting the same cytokines differentially in both compartments. There is also evidence that fluid balance in the lungs is regulated by active ion transport which is the likely mechanism for removal of alveolar oedema.85 The majority of patients with acute lung injury have impaired alveolar epithelial fluid transport, which is associated with a higher mortality. Under pathological conditions, these mechanisms may also influence movement of inflammatory mediators from the lungs to the systemic circulation, although further studies will be necessary to explore this. Third, protein concentration is difficult to quantify in airway secretions. This fluid is mobile in the airway moving upwards, is variably hydrated and usually contaminated during the collection process (lavage or expectoration). This results in highly variable dilution and the repeated measurements of inflammatory indices in the airways have marked intra- and inter-patient variability.81 Furthermore, proteins produced in the interstitial space may not be measured in airway secretions. Thus, even if the plasma level is constant, correlations will be difficult to identify with these factors influencing the potential source. Sequential sampling and averaging the data for an individual dramatically reduces this variability.81 Nevertheless, even with this methodology, direct measurements of TNFα and IL-1 in the secretions and plasma show no direct correlation.83
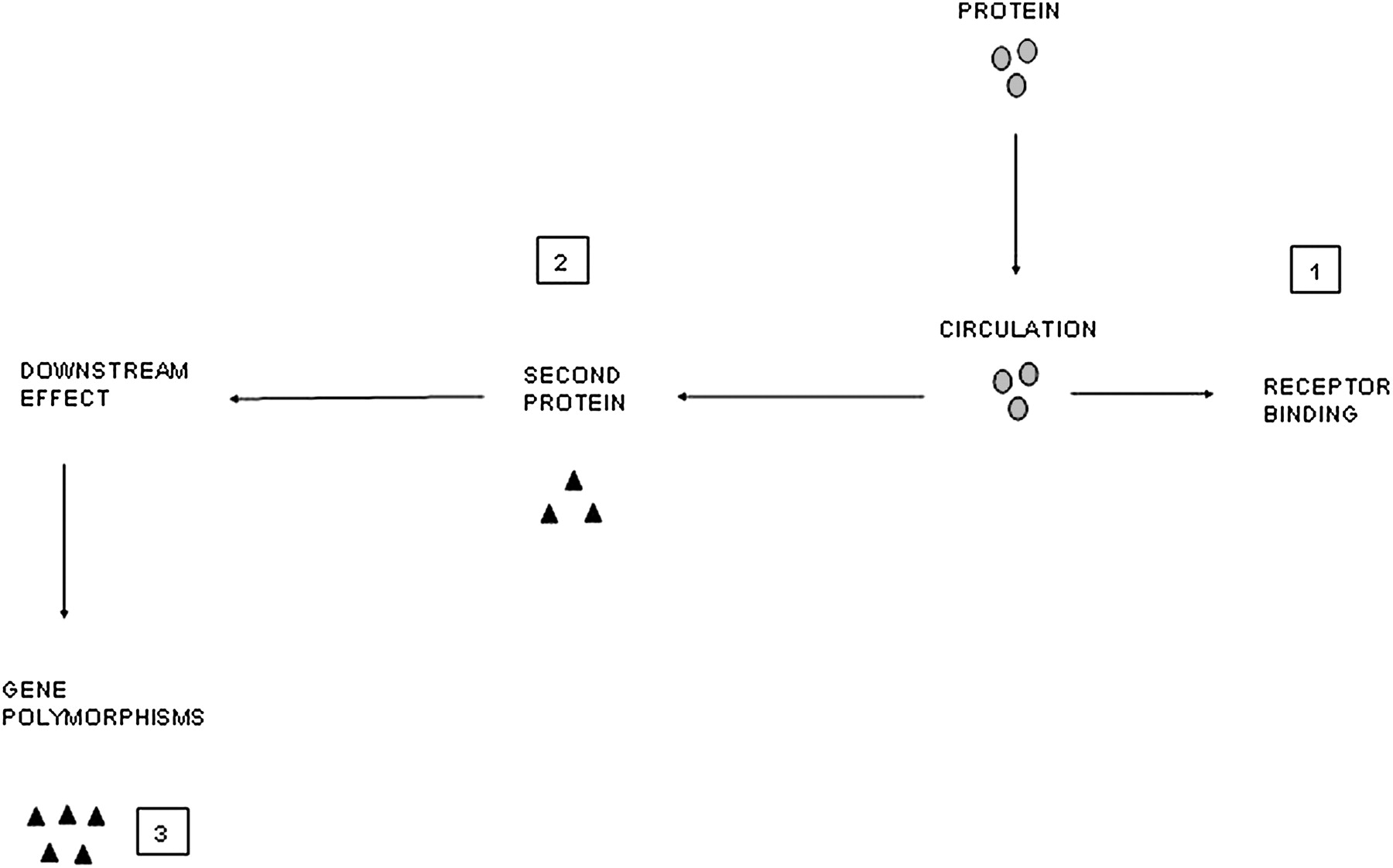
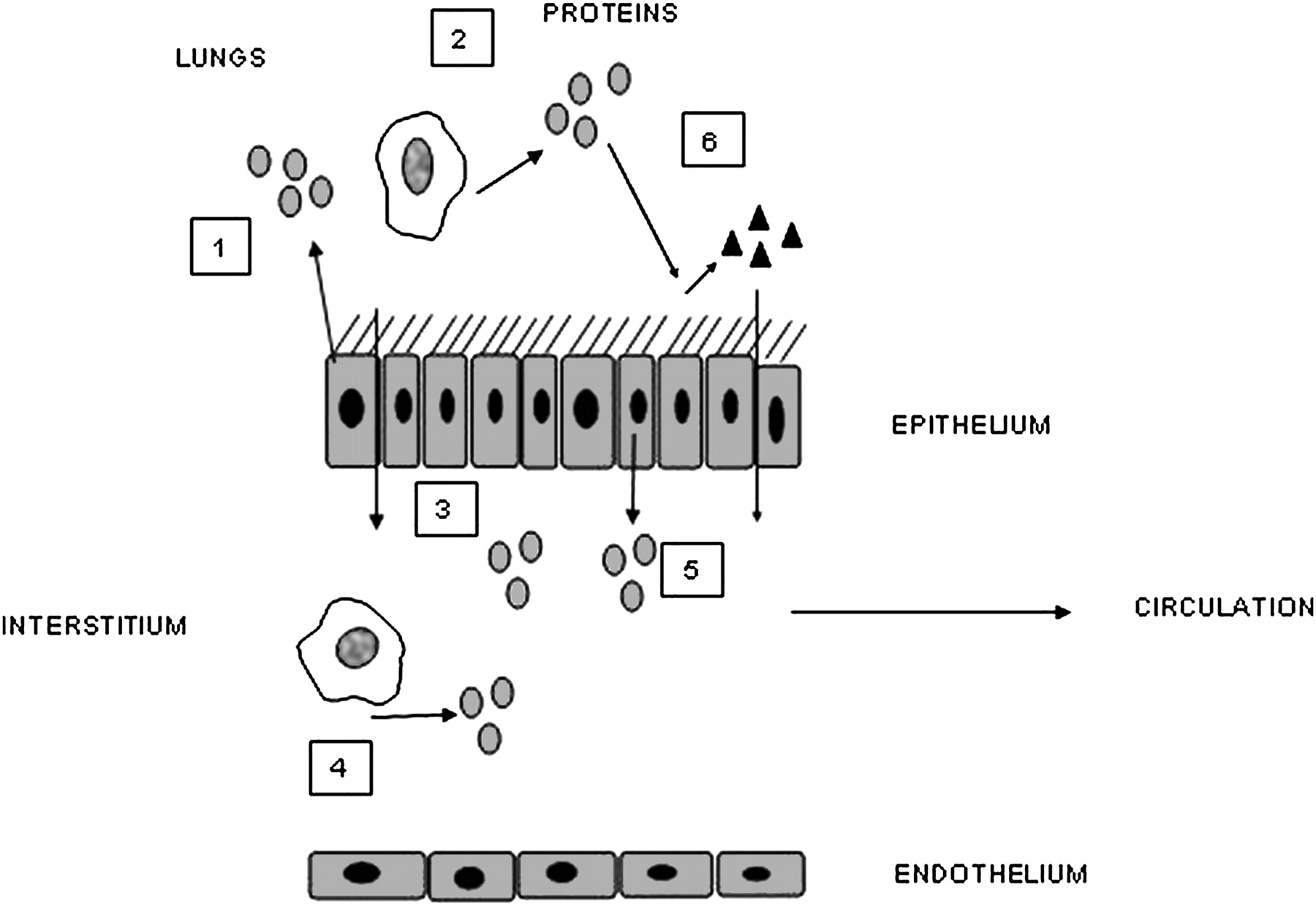
However, there are other complexities that will influence the result. Local release of cytokine A may lead to a local autocrine or paracrine effect, releasing cytokine B either into the same compartment or a different one in the lung, and it may be cytokine B that has the systemic effect. The volume of distribution of the cytokine is markedly different and receptor binding will influence measurement of free cytokine. In the lung, receptors are often saturated in the presence of marked inflammation leaving a surplus of free (and hence measurable) cytokine. This is unlikely in plasma where there are abundant circulating and tissue receptors resulting in rapid uptake of any leaked cytokine. This process may then result in a downstream ‘overspill’ effect with receptor binding of cytokine A stimulating systemic production of factor B (eg, IL-6 release leading to CRP production).86 Even this process itself does not represent a straight line effect. Polymorphisms of the CRP gene will result in differential responses to IL-685 and this may be important in cardiovascular mortality.87 These concepts are summarised in figures 1 and 2.

Diagrammatic representation of mechanisms that influence the process of ‘overspill’ and its detection. Several processes will influence the release of lung cytokines into the circulation. (1) Cytokines are released from bronchial epithelial cells into lung secretions. (2) Other inflammatory cells may also release the same cytokines into the lung secretions. (3) These cytokines may move into the interstitium by diffusion depending on size and inflammation. (4) Migrating monocytes, tissue macrophages or other inflammatory cells may release cytokines directly into the interstitial space. These proteins may not necessarily be present in airway secretions. (5) Protein movement may be facilitated across the epithelial cells or secreted basolaterally. The proteins in the interstitium can then enter the lymph and hence systemic circulation. (6) Local release of a cytokine may lead to downstream production of a second cytokine locally or in the interstitium with subsequent passage into the circulation.
{kind=link}
{kind=link}
Diagrammatic representation of mechanisms that influence the process of ‘overspill’ and its detection. Once in the circulation, other mechanisms will influence the measurement and downstream effects of proteins. (1) Receptor binding influences measurement of free cytokines. (2) Systemic release of one cytokine may lead to a downstream effect and the release of a second protein. It may be the second protein that is the systemic signal (eg, interleukin 6 leading to C-reactive protein production). (3) Gene polymorphisms exist which may affect the response of the downstream marker to any given cytokine leading to differential production.
These complexities make it generally unlikely that direct evidence (correlation between lung and plasma cytokine concentrations) for ‘overspill’ will be detected. At best we are left with inflammation relationships, and further extensive studies will be needed to confirm or indeed refute the concept of ‘overspill.’
Conclusion
There is direct evidence from α1-antitrypsin and secretory leukoprotease inhibitor that protein movement occurs from the lungs to lymph and blood. SPD (which is predominantly produced in the lung) is elevated in the serum of patients with COPD and its concentration relates to disease severity and symptoms, providing further evidence for leaking of proteins from the lung to the systemic circulation. Evidence from inhaled substances such as air pollutants and cigarette smoke has demonstrated a temporal link between the inflammatory process in the lung and systemic inflammation. Experimental data indicate that AM and bronchial epithelial cells are critically important in processing inhaled noxious gases and particles, and that the mediators they produce are also identified in the systemic response in COPD. Studies have also consistently shown alterations in circulating inflammatory cells in COPD such as neutrophils and lymphocytes, indicating a downstream effect. Clinical effects of lung inflammation are clearly implied by correlations between secretion cytokine concentrations and BMI and the mechanisms of several comorbidities.
Proteins originating from the lung may have both direct and indirect effects systemically. A lack of correlation between lung and systemic concentrations of any single mediator does not mean that ‘overspill’ has not occurred. Proving the concept of ‘overspill’ has been difficult due to underpowered studies, methods of quantifying protein levels and a lack of awareness of the complex processes that affect the measurement or response. Clearly, further studies in this area are needed to confirm or refute the ‘overspill’ hypothesis.
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.