Article Text

Abstract
Background: Hypercapnic acidosis exerts protective effects in acute lung injury but may also slow cellular repair. These effects may be mediated via inhibition of nuclear factor-κB (NF-κB), a pivotal transcriptional regulator in inflammation and repair.
Objectives: To determine the effects of hypercapnic acidosis in pulmonary epithelial wound repair, to elucidate the role of NF-κB and to examine the mechanisms by which these effects are mediated.
Methods: Confluent small airway epithelial cell, human bronchial epithelial cell and type II alveolar A549 cell monolayers were subjected to wound injury under conditions of hypercapnic acidosis (pH 7.0, carbon dioxide tension (Pco2) 11 kPa) or normocapnia (pH 7.37, Pco2 5.5 kPa) and the rate of healing determined. Subsequent experiments investigated the role of hypercapnia versus acidosis and elucidated the role of NF-κB and mitogen-activated protein kinases. The roles of cellular mitosis versus migration and of matrix metalloproteinases in mediating these effects were then determined.
Results: Hypercapnic acidosis reduced wound closure (mean (SD) 33 (6.3)% vs 64 (5.9)%, p<0.01) and reduced activation of NF-κB compared with normocapnia. Buffering of the acidosis did not alter this inhibitory effect. Prior inhibition of NF-κB activation occluded the effect of hypercapnic acidosis. Inhibition of ERK, JNK and P38 did not modulate wound healing. Hypercapnic acidosis reduced epithelial cell migration but did not alter mitosis, and reduced matrix metalloproteinase-1 while increasing concentrations of tissue inhibitor of metalloproteinase-2.
Conclusions: Hypercapnic acidosis inhibits pulmonary epithelial wound healing by reducing cell migration via an NF-κB dependent mechanism that may involve alterations in matrix metalloproteinase activity.
Statistics from Altmetric.com
Recent clinical trials have shown that, in patients with acute respiratory distress syndrome (ARDS), outcome is improved when a “protective” ventilatory strategy is used.1 These ventilatory strategies minimise ventilator-induced lung injury (VILI) by reducing tidal volume and minute ventilation in order to limit the degree of lung stretch. The hypercapnic acidosis (HCA) that occurs in the setting of acute lung injury (ALI)/ARDS is a consequence both of these protective ventilatory strategies and of the disease itself which can limit carbon dioxide (CO2) clearance. While this hypercarbia has traditionally been considered a passive phenomenon, it is increasingly clear that HCA is a potent biological agent. HCA has been shown directly to attenuate ALI induced by free radicals,2 pulmonary3 and systemic ischaemia-reperfusion,4 pulmonary endotoxin instillation,5 severe evolving6 and established bacterial pneumonia7 and systemic sepsis.8 9 Of importance in the context of protective ventilation, HCA directly attenuates VILI.10 11 These protective actions of HCA appear to be due in part to its anti-inflammatory effects.12 13
The potential for HCA to modulate the repair process following injury is less well characterised. At a cellular level, HCA has been shown to delay plasma membrane resealing, a key cellular reparative mechanism.14 Excessive lung stretch also produces gross alveolar wound lesions due to the large shear forces produced, both in the clinical setting15 and in experimental VILI models.16 Repair of wounds in the pulmonary epithelial surface is therefore critical to the repair process following mechanical injuries such as VILI.
The anti-inflammatory effects of HCA may be mediated, at least in part, via inhibition of the nuclear factor kappa B (NF-κB) pathway,17 a key transcription factor during cell injury or stress. NF-κB is also an important cytoprotectant and plays a key role in mediating cell survival and wound repair in other epithelia.18 HCA-mediated inhibition of NF-κB activation could therefore retard pulmonary epithelial wound healing, slowing the recovery following VILI.
Matrix metalloproteinases (MMPs) are a family of enzymes that can degrade extracellular matrix proteins including basement membrane and tight junctions. Tissue inhibitors of metalloproteinases (TIMPs) exist, with TIMP-1 and TIMP-2 the major secreted TIMPs in the lung. MMPs have been implicated in the pathogenesis of ARDS, and may increase tissue breakdown and contribute to loss of the alveolo-capillary barrier and intercellular junctions.19 However, MMPs are also central to lung repair, with a favourable balance of MMPs to TIMPs necessary to facilitate cell detachment from the basement membrane and migration during wound healing.20 The production of several MMPs (including MMP-1) and of TIMP-1 and TIMP-2 is dependent on NF-κB.20
In view of these considerations, we have explored the effects and mechanisms of action of HCA on pulmonary epithelial wound repair. We sought to determine whether this effect of HCA was a function of the acidosis or hypercapnia per se, and to elucidate the role of NF-κB and mitogen-activated protein (MAP) kinases in this process. Subsequent experiments determined the contribution of altered migration versus cell replication and the role of MMPs and TIMPs in HCA-induced inhibition of pulmonary epithelial wound healing.
Methods
Primary small airway epithelial cells (SAECs), type II alveolar A549 cells and human bronchial epithelial (HBE) cells were used in all experiments. The SAEC primary cells, which are harvested human lung cells from healthy donors, were purchased from Lonza Walkersville (Walkersville, Maryland, USA) as cryopreserved passage 1 cultures and used at passages 3–5. A549 cells were purchased from the European collection of cell cultures (Porton Down, UK) as cryopreserved 90-passage cultures and used at passages 91–95. The 16HBE14o- (HBE) transformed epithelial cell line was a gift from D Gruender (University of Vermont, Burlington, Vermont, USA).21 All cells were then grown to confluence on plastic plates or tissue culture flasks of the required size (Corning, New York, USA) at 37°C in a humidified incubator saturated with a gas mixture containing 5% CO2 in air. Once the cells had grown to confluence, single or multiple wounds were made in each well by scraping off cells with a 200 μl pipette tip, as previously described.18 Pre-equilibrated and prewarmed media were then added to the wells and the plates were randomised to incubation in either normocapnia (Fico2 0.05, balance air) or HCA (Fico2 0.15, balance air) as per group allocation. At 24 h and/or 48 h later, the monolayers were fixed with 4% paraformaldehyde in phosphate-buffered saline (w/v) and stained with haematoxylin and eosin. The extent of epithelial restitution was determined by imaging each plate on a flatbed scanner and assessing the area of each wound using edge-finding software (Photoshop V.8.0, Adobe Systems, San Jose, California, USA). Additional details regarding these cell lines and the wound assessment technique are provided in the online data supplement.
Confluent SAEC, pulmonary alveolar A549 and HBE epithelial layers were each subject to single wound injury as described above and subsequently incubated under conditions of normocapnia (5% CO2, pH 7.37, carbon dioxide tension (Pco2) 5.5 kPa) or HCA (15% CO2, pH 7.0, Pco2 11 kPa). The dose-response characteristics of HCA were then determined. Confluent alveolar epithelial A549 cell cultures were incubated under conditions of normocapnia (5% CO2, pH 7.37, Pco2 5.5 kPa) and moderate (10% CO2, pH 7.15, Pco2 8 kPa) or more severe (15% CO2, pH 7.0, Pco2 11 kPa) HCA. The effect of hypercapnia versus acidosis on pulmonary epithelial healing was then determined. The RPMI-1640 medium, which includes 2.2 g/l sodium bicarbonate (NaHCO3) with 10% fetal bovine serum, was used in these experiments. In preliminary studies we determined that an additional 3.2 g/l NaHCO3 was required to buffer the pH of the medium back to 7.35–7.40 when incubated in 15% CO2, and to produce normocapnic alkalosis when cells are incubated in 5% CO2. An equiosmolar amount of sodium chloride was added to the cells exposed to normocapnia and HCA to eliminate any potential effect of differences in medium osmolarity on wound healing. This produced the following groups: normocapnia (pH 7.37, Pco2 5.5 kPa); metabolic alkalosis (pH 7.8, Pco2 5.5 kPa); HCA (pH 7.0, Pco2 11 kPa); and buffered hypercapnia (pH 7.37, Pco2 11 kPa). The effect of these conditions on the extent of wound healing was then determined. Additional experiments were carried out using the cell permeant pH sensitive fluorimetric dye carboxy SNARF-1, acetoxymethyl ester acetate (10 μM, Invitrogen, Carlsbad, California, USA) to determine the intracellular pH in the epithelial layers exposed to normocapnia, normocapnic alkalosis, HCA and buffered hypercapnia.
Subsequent experiments determined the effect of wound injury and HCA on NF-κB activation. A549 cells transfected with κB-luciferase and TK-renillin control reporter constructs were subjected to multiple wound injury and the extent of NF-κB activation determined under conditions of normocapnia or HCA. Western blotting was used to determine the degradation profile of IκBα in both uninjured and wounded A549 epithelial cultures under conditions of normocapnia and HCA. In addition, the extent of wound healing under HCA and normocapnia was determined in the presence of pharmacological (pyrollidine dithiocarbamate (PDTC) 90 μM, Sigma, Poole, UK; BAY 11-7085 10 μM, Calbiochem, San Diego, California, USA)) and gene overexpression (IκBα-SR “super-repressor”) mediated inhibition of NF-κB. The role of the MAP kinases ERK, JNK and P38 in mediating the effect of HCA was also determined using pharmacological inhibitors.
The contribution of alterations in cell replication and cell migration to the inhibitory effect of HCA was determined. Confluent A549 monolayers were incubated with the mitosis inhibitors busulfan (10 μM, Sigma) or indirubin-3′-monoximine (1 μM, Calbiochem) in the presence of normocapnia and HCA. Subsequently, A549 cells were seeded into each upper chamber of an Invasion Assay plate (R&D Systems, Minneapolis, Minnesota, USA) and the migration of these cells in response to the chemoattractant cytokines interleukin 8 (IL8) and monocyte chemotactic protein 1 (MCP-1) and the pro-inflammatory cytokine tumour necrosis factor α (TNFα) was determined under conditions of normocapnia and HCA. The chemotactic potential of conditioned medium from wounded A549 cultures incubated under conditions of normocapnia or HCA was then determined. Finally, the concentrations of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) were measured in cell culture supernatants from epithelia subjected to multiple wounds under conditions of normocapnia and HCA, as previously reported.22
Data analysis
Results are expressed as mean (SD) for normally distributed data and as median (interquartile range) if non-normally distributed. Data were analysed by t test, one-way ANOVA or Kruskal–Wallis followed by the Mann–Whitney U test with the Bonferroni correction for multiple comparisons, as appropriate. A p value of <0.05 was considered statistically significant.
Results
HCA inhibits pulmonary epithelial wound healing
HCA reduced the rate of wound closure in primary SAEC cultures 24 h after wound injury compared with normocapnia (fig 1A). This inhibition of pulmonary epithelial wound closure was also seen in alveolar epithelial A549 cell monolayers at 24 h and 48 h following wound injury compared with control conditions (33 (6.3)% vs 64 (5.9)%, p<0.01; fig 1B). In subsequent similar experiments, HCA also reduced the rate of wound closure in HBE cell monolayers (see fig A in online supplement), demonstrating that this effect of HCA occurs in multiple pulmonary epithelial cell types. HCA had no effect on epithelial cell morphology in the 48 h following wound injury (see fig B in online supplement). The effect of HCA on the rate of wound closure was dose-dependent. Moderate degrees of HCA (10% CO2, pH 7.15, Pco2 8 kPa) reduced wound closure in A549 epithelial layers over 48 h compared with normocapnia. This inhibition of wound closure was significantly less than that seen with exposure to more severe HCA (15% CO2, pH 6.9, Pco2 11 kPa; fig 1C).

(A) Hypercapnic acidosis (HCA) reduced wound closure in the first 24 h following scrape injury in primary lung small airway epithelial (SAEC) cell layers in comparison with normocapnic conditions. (B) HCA reduced the rate of wound closure at both 24 and 48 h following scrape injury in pulmonary alveolar (A549) cell epithelial layers in comparison with normocapnic conditions. (C) The effect of HCA on the rate of wound closure was dose-dependent. Incubation of A549 epithelial layers under conditions of moderate HCA (10% CO2, pH 7.15, carbon dioxide tension (Pco2) 8 kPa) reduced wound closure over 48 h compared with normocapnia, but to a significantly lesser extent than that seen with exposure to more severe HCA (15% CO2, pH 7.0, Pco2 11 kPa). (D) The effect of HCA on wound healing appeared to be a function of the hypercapnia rather than the acidosis per se. A549 epithelial cell layers were exposed to HCA, buffered hypercapnia, normocapnia or metabolic alkalosis, subjected to scrape wounding and the extent of restitution of the wound determined over 24 h. There was no difference in the extent of wound healing in epithelial layers exposed to buffered hypercapnia compared with HCA. In contrast, normocapnic alkalosis did not affect the rate of wound healing compared with normocapnia. *p<0.05 vs normocapnia.
Buffered hypercapnia inhibits pulmonary epithelial wound healing
Buffered hypercapnia retarded wound repair in A549 alveolar epithelial cell monolayers. The degree of inhibition of wound closure was not different from that seen with HCA (fig 1D). Furthermore, normocapnic alkalosis did not alter wound repair compared with normocapnia. Additional experiments using the cell permeant pH sensitive fluorimetric dye SNARF-1 to determine intracellular pH showed that the intracellular pH in the epithelial layers exposed to buffered hypercapnia was not different from that seen with normocapnia (see fig C in online supplement). These findings indicate that the effect of HCA is a function of the hypercapnia rather than pH per se.
HCA inhibits wound healing via reduced NF-κB activation
HCA reduced the activation of NF-κB following wound injury to the pulmonary epithelium. The transcription of a κB-luciferase construct in A549 epithelial layers following wound injury was significantly reduced with HCA compared with normocapnia (fig 2A). Direct inhibition of NF-κB activation with PDTC and BAY 11-7085 significantly inhibited wound healing under conditions of normocapnia (fig 2B). The degree of inhibition seen with PDTC and BAY 11-7085 was similar to that with HCA. HCA did not further inhibit wound healing in the presence of PDTC or BAY 11-7085, demonstrating that the effect of HCA was completely occluded by direct inhibition of NF-κB.

(A) Hypercapnic acidosis (HCA) reduced the activation of nuclear factor kappa B (NF-κB) following wound injury to the pulmonary epithelium. The transcription of a κB-luciferase construct in A549 epithelial layers following wound injury was significantly reduced with HCA compared with normocapnia. (B) Direct inhibition of NF-κB by pyrollidine dithiocarbamate (PDTC) and BAY 11-7085 attenuated wound healing in A549 monolayers and occluded the effect of HCA. (C) Following transduction with an adeno-associated virus construct encoding the IκBα transgene or vehicle, human bronchial epithelial (HBE) monolayers were grown to confluence, subjected to scrape wounding and exposed to normocapnia or HCA. Cells were then fixed at baseline (time 0) and 24 h and the extent of restitution of the wound determined. (D) HCA reduced the degradation of IκBα following wound injury compared with normocapnia. HBE monolayers were subject to multiple wound scraping and exposed to normocapnia or HCA. Cells were then harvested at 0, 60 and 180 min and examined for IκBα degradation by Western blotting. *p<0.05 versus all other groups (Student–Newman–Keuls test following one-way ANOVA).
Overexpression of the IκBα transgene also inhibited wound healing under conditions of normocapnia (fig 2C). The extent of inhibition was similar to that seen with HCA. There was some further inhibition of wound healing by HCA in the presence of IκBα overexpression, suggesting incomplete NF-κB blockade by IκBα.
A Western blot of protein from HBE cell cultures demonstrated degradation of IκBα, a key event in the propagation of the NF-κB response, at 60 min following wound injury. HCA inhibited this degradation when compared with normocapnic controls (fig 2D).
Effect of HCA is independent of MAP kinase and mitosis
The effect of HCA on wound healing was independent of MAP kinases. Inhibition of the MAP kinases ERK, JNK and P38 did not attenuate wound healing in the presence or absence of HCA. Interestingly, inhibition of both ERK and JNK induced a small but statistically significant increase in wound healing. This effect of JNK was seen in the presence and absence of HCA and was therefore independent of HCA (see fig D in online supplement).
Pharmacological inhibition of mitosis with busulfan inhibited wound healing in the setting of both normocapnia and HCA. The mitosis inhibitor indirubin-3′-monoximine also inhibited wound healing in the presence of HCA (see fig E in online supplement). Taken together, these findings indicate that the effects of HCA on wound healing are not mediated via inhibition of mitosis.
HCA reduces cell migration
Incubation of alveolar A549 cells under conditions of HCA inhibited cell migration in both the presence and absence of a chemotactic stimulus compared with normocapnia. This inhibition of cell migration by HCA persisted at each concentration of the IL8, MCP-1 and TNFα cytokine combination tested (fig 3A).

(A) Hypercapnic acidosis (HCA) inhibited pulmonary epithelial cell migration in response to a chemotactic stimulus. A549 cells were seeded into the upper chamber of a collagen type I coated invasion assay plate. HCA inhibited cell migration at all concentrations of a combination of tumour necrosis factor α, monocyte chemotactic protein 1 and interleukin 8 added to the lower chamber. (B) HCA reduced the concentration of chemotaxins in media from wounded and uninjured epithelia. The migration of A549 cells towards conditioned medium from wounded and uninjured epithelial layers exposed to HCA was reduced compared with migration towards medium from wounded and uninjured epithelial cultures exposed to normocapnia. The values for migration in both experiments are normalised to migration towards serum-free fresh medium. *p<0.05 vs normocapnia. No wound indicates cell culture media from uninjured A549 cultures. Wound indicates media from wound injured A549 cultures.
The conditioned medium from A549 monolayers subjected to wound injury and incubated under conditions of HCA reduced migration of A549 cells compared with normocapnia (fig 3B). This reduction in cellular migration by HCA was also seen with conditioned media from A549 cells not subject to wound injury. These findings suggest that A549 cells exposed to HCA may produce lower concentrations of factors that promote migration and/or higher concentrations of factors that inhibit cellular migration.
HCA modulates MMP and TIMP concentrations
Conditioned medium from A549 monolayers subjected to wound injury under both normocapnia and HCA conditions were assayed for MMP-1, MMP-3 and MMP-7 and for TIMP-1 and TIMP-2. These MMPs and TIMPs are central to cellular migration and wound healing. The concentration of MMP-1 in the medium was significantly reduced in wounded epithelial layers incubated in HCA compared with normocapnia (fig 4A). The concentration of MMP-7 was also reduced by HCA, but this difference was not statistically significant (fig 4B). MMP-3 was not detected in measurable quantities in the medium.
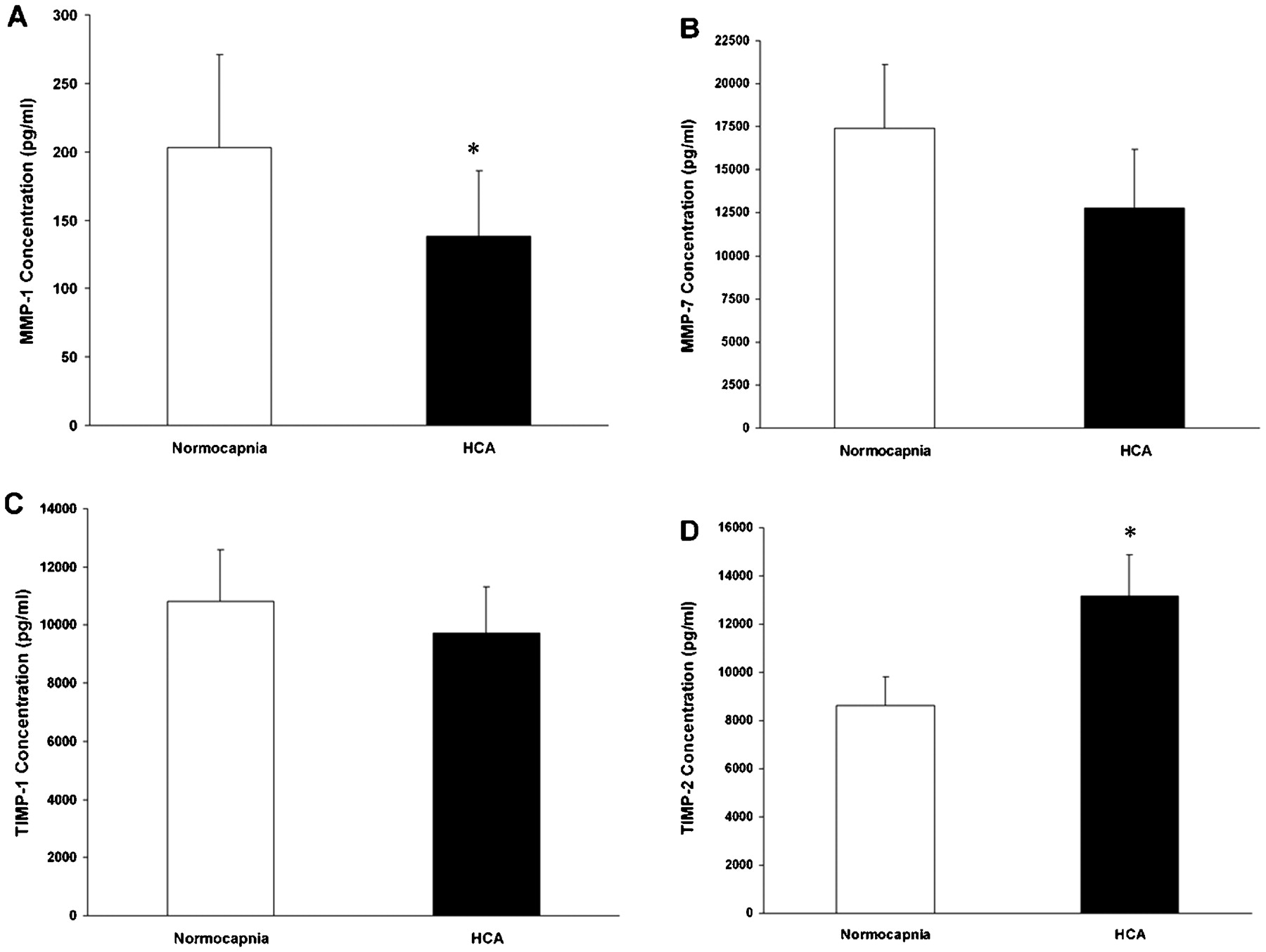
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Hypercapnic acidosis (HCA) significantly reduced the concentration of matrix metalloproteinase-1 (MMP-1) in the medium from A549 epithelial layers subjected to wound injury in comparison with wounded epithelia exposed to normocapnia. (B) The concentration of MMP-7 in the medium from A549 epithelial layers subjected to wound injury under HCA conditions was not different from that in wounded epithelia exposed to normocapnia. (C) The concentration of tissue inhibitor of metalloproteinases 1 (TIMP-1) in the medium from A549 epithelial layers subjected to wound injury under HCA conditions was not different from that in wounded epithelia exposed to normocapnia. (D) HCA significantly reduced the concentration of TIMP-2 in the medium from A549 epithelial layers subjected to wound injury in comparison with wounded epithelia exposed to normocapnia. *p<0.05 vs normocapnia (t test).
The concentration of TIMP-1 in the medium of scrape wounded cells was not altered by incubation in HCA (fig 4C). In contrast, the concentration of TIMP-2 in the medium was significantly increased in scrape wounded cells incubated in HCA compared with normocapnia (fig 4D).
Discussion
We have shown for the first time that HCA impairs pulmonary epithelial wound healing. The mechanisms underlying this effect of HCA appear to be NF-κB dependent. The impairment of wound healing involves inhibition of cellular migration rather than mitosis and is independent of MAP kinase. The underlying mechanisms involve HCA-induced alterations in the balance between MMPs and their tissue inhibitors. The relevance of these findings is clear, given the prevalence of hypercapnia in patients managed with protective lung ventilation12 13 and the fact that pulmonary epithelial wounds, which result from mechanical disruption, are a key component of VILI.15 16
Risks and benefits of HCA in protective lung ventilation
We have proposed a new paradigm—“therapeutic hypercapnia”—wherein the increased CO2 might contribute to lung protection.12 To explore this, we established several ALI models and have demonstrated protective effects in ALI induced by free radicals,2 23 endotoxin,5 both primary3 and secondary4 ischaemia-reperfusion, high lung stretch,10 severe evolving6 and established bacterial pneumonia7 and systemic sepsis.9 Key mechanisms by which HCA exerts beneficial effects include attenuation of free radical injury,2 apoptosis,3 nitric oxide-derived oxidant generation5 and NF-κB activation.17 In the clinical setting, multivariate analysis of data from the ARDSnet trial1 suggests that HCA increased survival in patients randomised to high tidal volume ventilation.24
More recently, our group and others have questioned the safety of HCA.13 25 HCA may potentiate protein nitration by peroxynitrite, a potent free radical, causing these proteins to malfunction.26 We have recently reported that HCA impairs bacterial killing in the setting of prolonged lung infection, ultimately worsening ALI.27 HCA may also delay healing following ALI/ARDS. HCA delays plasma membrane resealing following VILI, a key reparative mechanism following ALI/ARDS.14 HCA may therefore constitute a double-edged sword, attenuating injury pathways2 3 4 5 10 11 23 but retarding the response to pulmonary bacterial sepsis27 28 and slowing lung repair.14
NF-κB: a key transcriptional regulator
The NF-κB protein complex consists of RelA (which is involved in dimerisation, DNA binding and transcriptional regulation) and P50 or P65 homo- or heterodimers. NF-κB is normally sequestrated in the cytoplasm of non-stimulated cells, bound to IκB inhibitory proteins. NF-κB is activated by stimuli such as lipopolysaccharide, pro-inflammatory cytokines and viral protein via diverse signalling pathways that converge to phosphorylate IκB kinase complex proteins (IKK). IKK then phosphorylates IκB proteins on two N-terminal serine residues, which then dissociates from NF-κB and is degraded. NF-κB then translocates to the nucleus and binds to specific cognate binding sequences in the promoter or enhancer regions of different inflammatory target genes where it initiates the transcriptional apparatus. NF-κB regulates genes central to lung injury, inflammation and repair, and is a key therapeutic target in ALI/ARDS.29 30 Inhibition of NF-κB is protective in diverse models of inflammatory ALI.29 31 32 However, NF-κB is cytoprotective, promoting cell survival and wound repair18 and resistance to infection.33
Effects and mechanisms of action of HCA in epithelial wound repair
Our findings indicate that HCA potently reduces the rate of pulmonary epithelial wound closure. This effect of HCA appears to be a function of the hypercapnia rather than the acidosis per se. In fact, buffered hypercapnia retarded wound repair to an extent similar to that seen with HCA. The intracellular pH in buffered hypercapnia cultures was the same as that in epithelial cultures exposed to normocapnia. Normocapnic alkalosis did not modulate wound healing, which further illustrates that pH per se did not modulate wound healing in this model. In contrast, the effect of HCA to reduce pulmonary epithelial cell membrane resealing, a related but distinct component of pulmonary epithelial repair, is attenuated by buffering.34 In addition, the beneficial effects of HCA on lung permeability in vitro are abolished by buffering.23 Therefore, buffering appears to abolish the protective effects of HCA but not its effect on restitution of the pulmonary epithelium.
The effects of HCA on pulmonary epithelial wound repair appear to be mediated via activation of NF-κB. Both PDTC and BAY 11-7085, which are pharmacological NF-κB inhibitors, directly retarded wound healing but did not have any effect in the presence of HCA. This suggests that these inhibitors shared a mechanism of action with HCA. This is supported by the finding that overexpression of the IκBα transgene did not modulate wound healing in the presence of HCA. The effects of HCA on pulmonary epithelial wound healing appear to be MAP kinase independent. Pharmacological inhibition of mitosis did not differentiate modulate wound healing in the setting of normocapnia versus HCA, suggesting that the generation of new cells is not central to the mechanism of healing in this model. This finding is consistent with previous studies that report that cell replication does not play a role in restitution of pulmonary epithelial wounds.35
In contrast, migration of cells in response to inflammatory cytokines and media from wounded cell cultures was impaired by HCA. This finding suggests that HCA reduces wound healing by a process involving decreased cell migration. This is consistent with previous studies reporting that cell migration is central to restitution of pulmonary epithelial wounds.36 HCA significantly reduced the concentrations of MMP-1 and increased TIMP-2 in the supernatant from wounded cell cultures. MMP-7—which is derived specifically from epithelial cells—was also decreased, although this did not reach statistical significance. Overall, this alteration in MMP/TIMP balance provides a mechanism for the reduced cellular migration potential induced by HCA. Furthermore, MMP-1, TIMP-1 and TIMP-2 are all NF-κB dependent,20 which provides a potential mechanism by which HCA-mediated NF-κB inhibition results in decreased cell migration and reduced wound healing.
Clinical significance
Doerr et al showed that HCA reduced the rate of plasma membrane repair, an important component of the cellular repair process following VILI.14 In this study, we have shown that HCA also reduces repair of gross pulmonary epithelial wounds, similar to those seen following high stretch ventilation in patients with ARDS.16 Ultrastructural studies in models of VILI confirm that wounds of this type are commonly found in the lung epithelium following high stretch ventilation,15 particularly at high capillary pressures.15 As our experiments were conducted in pulmonary epithelial cell cultures, caution is warranted before extrapolating to the clinical context. Our findings should be seen as hypothesis-generating and will need to be confirmed in further studies in preclinical models. Nevertheless, when taken together with the findings of Doerr et al, our studies raise clear concerns regarding the potential for HCA to delay repair of the pulmonary epithelium following mechanical injury, such as is seen in patients with ALI/ARDS undergoing mechanical ventilatory support. These findings may also be of relevance to repair in other organs in the setting of hypercapnia, and in other lung diseases where hypercapnia is prevalent such as chronic obstructive airways disease.
Conclusions
We have demonstrated for the first time that HCA impairs pulmonary epithelial wound healing. The mechanism underlying this effect of HCA is NF-κB dependent, involves inhibition of cellular migration rather than mitosis, which may be mediated by modification of MMP/TIMP balance, and is MAP kinase independent. These findings suggest that HCA may constitute a double-edged sword in ALI/ARDS, attenuating injury pathways but slowing lung repair.
REFERENCES
Supplementary materials
web only appendices
Files in this Data Supplement:
Footnotes
▸ Additional details and figures are published online only at http://thorax.bmj.com/content/vol64/issue11
This work was presented in part at the American Thoracic Society annual conference, Toronto, May 2008.
Funding This study was funded by the Health Research Board, Ireland (DO’T, BH, JGL) under the project grant award RP-2005-244.
Competing interests None.
DO’T and PH contributed equally to this paper.
Provenance and Peer review Not commissioned; externally peer reviewed.