Article Text

Abstract
Background Lymphadenitis is the most common extrapulmonary tuberculosis (EPTB) manifestation. The microbiome is important to human health but uninvestigated in EPTB. We profiled the site-of-disease lymph node microbiome in tuberculosis lymphadenitis (TBL).
Methods Fine-needle aspiration biopsies were collected from 158 pretreatment presumptive TBL patients in Cape Town, South Africa. 16S Illumina MiSeq rRNA gene sequencing was done.
Results We analysed 89 definite TBLs (dTBLs) and 61 non-TBLs (nTBLs), which had similar α- but different β-diversities (p=0.001). Clustering identified five lymphotypes prior to TB status stratification: Mycobacterium-dominant, Prevotella-dominant and Streptococcus-dominant lymphotypes were more frequent in dTBLs whereas a Corynebacterium-dominant lymphotype and a fifth lymphotype (no dominant taxon) were more frequent in nTBLs. When restricted to dTBLs, clustering identified a Mycobacterium-dominant lymphotype with low α-diversity and non-Mycobacterium-dominated lymphotypes (termed Prevotella-Corynebacterium, Prevotella-Streptococcus). The Mycobacterium dTBL lymphotype was associated with HIV-positivity and features characteristic of severe lymphadenitis (eg, larger nodes). dTBL microbial communities were enriched with potentially proinflammatory microbial short-chain fatty acid metabolic pathways (propanoate, butanoate) vs nTBLs. 11% (7/61) of nTBLs had Mycobacterium reads BLAST-confirmed as Mycobacterium tuberculosis complex.
Conclusions TBL at the site-of-disease is not microbially homogeneous. Distinct microbial community clusters exist that, in our setting, are associated with different clinical characteristics, and immunomodulatory potentials. Non-Mycobacterium-dominated dTBL lymphotypes, which contain taxa potentially targeted by TB treatment, were associated with milder, potentially earlier stage disease. These investigations lay foundations for studying the microbiome’s role in lymphatic TB. The long-term clinical significance of these lymphotypes requires prospective validation.
- Tuberculosis
Data availability statement
Data are available on reasonable request. Data are available on reasonable request. Deidentified patient data, the study protocol, informed consent and datasets generated in this study may be requested from the corresponding author. Sequencing data reported in this paper will be released and made publicly available for sharing on publication (NCBI Bioproject PRJNA738676).
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Lymphadenitis is the most frequent extrapulmonary tuberculosis manifestation. The microbiome is critical for human health, however, the microbiome at the site-of-disease in patients with tuberculosis lymphadenitis is completely uncharacterised, including whether distinct microbial clusters (which we term ‘lymphotypes’) are associated with clinically important patient characteristics.
WHAT THIS STUDY ADDS
Surprisingly, patients with confirmed tuberculosis lymphadenitis often had bacterial taxa other than Mycobacterium dominant at the site-of-disease (Prevotella, Streptococcus, Corynebacterium). Such patients had milder forms of disease (eg, less swelling, less HIV) whereas patients with the Mycobacterium-dominated lymphotype had increased microbial functional capacity for proinflammatory short-chain fatty acids and more severe disease.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our findings have relevance for clinical staging and treatment of tuberculosis lymphadenitis, which we show to not be microbially homogeneous, and suggest that the site-of-disease in tuberculosis lymphadenitis is, prior to shifting to becoming Mycobacterium-dominated, first characterised by Prevotella, Streptococcus and/or Corynebacterium dominance and milder disease. Lastly, given that Streptococcus and Corynebacterium are themselves capable of causing lymphadenitis and susceptible to first-line TB treatment, such treatment may alleviate pathology in tuberculosis lymphadenitis by, in part, killing taxa other than Mycobacterium.
Introduction
Tuberculosis (TB), which kills 1.5 million people globally each year (including 214 000 people with HIV), causes extrapulmonary TB (EPTB).1 EPTB accounts for ~16% of all TB, up to half of all TB in people living with HIV (PLHIV)2 and has high mortality.
TB lymphadenitis (TBL) is the most common EPTB manifestation, accounting for 70% of EPTB and most frequently affects peripheral and cervical lymph nodes.3 4 TBL occurs after Mycobacterium tuberculosis (Mtb) enters the airways, is taken up by phagocytic cells, and transported to lymph nodes where granulomas may form. These steps are also necessary for priming T-cells to generate adaptive immune responses for microbial killing mediated by cytokines and other effector mechanisms.5
Lymph nodes have an important role in TB pathogenesis: enlargement has been documented following exposure, even if only a fraction of patients with enlarged nodes develop active disease.6 Animal studies show lymph nodes can be sites of TB reactivation (Mtb DNA found in new lung granulomas share unique DNA barcodes with Mtb previously only found in lymph nodes).7 Furthermore, pathologically normal lymph nodes obtained at autopsy from humans without active TB can, when used to inoculate animals, cause active disease,8 suggesting these lymph nodes contained live Mtb (and hence Mtb DNA). Lymph nodes are therefore hypothesised to serve as a Mtb growth and persistence niche6 that can spread to bodily sites9 (in animals lymph node infection almost always accompanies infection in the lungs7; suggesting that TB may primarily be a lymphatic rather than pulmonary disease.10 For example, the lymph nodes of participants with subclinical TB pathology demonstrate enhanced metabolic activity on positron emission tomography (PET)-CT scans.11 Together these studies show that lymph nodes have an important role in TB pathogenesis, however, the determinants of why Mtb sometimes successfully establishes itself in the lymph nodes and subsequently proliferates, including the potential role of other microbes, is understudied. Key to understanding this is characterising the local site-of-disease.
The microbiota modulates immune responses via microbially-derived metabolites known as short-chain fatty acids (SCFAs).12 Enriched pulmonary SCFAs predict TB risk in HIV-infected individuals stable on ART, and ex vivo addition of butyrate inhibits Mtb-induced proinflammatory responses.13 Two studies assessed lymph node microbial content,14 15 both in mesenteric lymph nodes in Crohn’s disease where reduced diversity was observed. The site-of-disease microbiome in TB is underexamined16: in bronchoalveolar lavage fluid (BALF), active pulmonary TB was associated with Mycobacterium enrichment and Streptococcus depletion.17 18
The site-of-disease microbiome in TBL (including in HIV-endemic settings where TB is common) remains uncharacterised. Therefore, given the apparent role of the lymph nodes in TB pathogenesis, and the importance of the microbiome as a modulator of immunity, we characterised the site-of-disease lymph microbiome in presumptive TBL patients from a high HIV burden setting19 before the potentially confounding effects of antibiotic-based TB treatment.
Methods
Patient recruitment and follow-up
Presumptive TBL participants (≥18 years) were recruited from Tygerberg Academic Hospital in Cape Town, SA (25 January 2017–11 December 2018). Participants were programmatically referred for a routine fine needle aspiration biopsy (FNAB) via the skin for the investigation of lymphadenopathy as described.19 Eligible participants were not on TB treatment within 6 months. Clinical and demographic data were collected by interview and medical record review. Patients programmatically diagnosed with TBLwere initiated on treatment, and study staff assessed treatment response by telephonic follow-up ≥12 weeks. The study had no role in patient management.
Specimen collection and processing
For each patient, two background DNA sampling controls were collected in microcentrifuge tubes prior to lymph node aspiration: a skin swab (collected into saline; Ysterplaat Medical Supplies, Cape Town, South Africa) of the site to be punctured, followed by a saline flush of the syringe to be used for aspiration. Aspiration and microbiological procedures are in online supplemental methods. Aspirated material from the third pass was collected into 500 µL sterile saline and stored at −80°C until batched DNA extraction.
Supplemental material
Routine specimen testing
Patients were categorised based on lymphatic or non-lymphatic mycobacteriological evidence, provided by the government programmatic laboratory (National Health Laboratory Service), and/or clinical decision to start treatment by the responsible clinician thereafter.
Case definitions
Briefly, definite-TBLs (dTBLs) had at least one Mtb complex (MTBC)-positive extrapulmonary or pulmonary specimen by Xpert or culture (figure 1). Alternatively, they had site-of-disease cytology compatible with active TB. Probable-TBLs (pTBLs) did not meet dTBL criteria but commenced treatment empirically. Non-TBLs (nTBLs) had no microbiological or cytological evidence of TB. Further detail is in online supplemental table S1.

Study flow chart. Fine-needle aspirates, skin and saline controls were collected from presumptive TBL patients. dTBLs, definite-TBL; MGIT960 culture, mycobacteria growth indicator tube 960 liquid culture; nTBLs, non-TBLs; pTBLs, probable TBLs; Smear: Smear microscopy; Ultra: Xpert MTB/RIF Ultra; Xpert: Xpert MTB/RIF.
Microbial DNA extraction and sequencing
DNA was extracted from specimens and controls using the PureLink Microbiome DNA Purification Kit (Invitrogen, Carlsbad, USA). The 16S rRNA gene V4 hypervariable region (150 bp read length) was amplified and sequenced (paired-ends) on the Illumina MiSeq platform. Lymph, skin swab and one in five saline flushes were extracted and sequenced.
Microbiome data analysis
16S rRNA gene sequences were processed, denoised and analysed in Quantitative Insights Into Microbial Ecology (QIIME 2, v2020.8)20 and DADA221 using closed-reference picking by assigning taxonomy at a 97% similarity against representative sequences in Greengenes (V.13.8).22 QIIME2 outputs (phylogenetic tree, feature table, taxonomy) and metadata were imported into R (V.3.5.2) and analyses done using phyloseq.23 Shannon’s index was calculated with vegan24 as measure α-diversity (within-sample diversity). Bray-Curtis distances were calculated as a measure of β-diversity (between-sample diversity) and were visualised as principal coordinate analysis plots. Dirichlet-Multinomial Mixtures (DMM) modelling was done to estimate the optimal number of clusters based on microbial compositional similarity.25 These clusters are herewith referred to as ‘lymphotypes’.
Inferred metagenome
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) V.2.1.3-b26 was used to predict gene family abundance with PICRUSt2 default options (picrust2_pipeline.py). The resulting gene table was mapped against the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database, and pathway abundances were inferred from predicted KEGG ORTHOLOGY (KO) abundances.
Differential abundance analysis
Differentially abundant taxa and pathways were identified using DESeq2 (V.1.22.2), which internally corrects and normalises data.27 Feature tables were pruned to have a mean relative abundance ≥5% in 0.5% of samples.28 DESeq2 was run on PICRUSt outputs to identify common pathways in oL4 versus each lymphotype (overall patients), and dL3 versus each other lymphotype (dTBLs). DESeq2 outputs with abundances and significance values for each discriminatory taxon and pathway were obtained (see online supplemental material: DESeq2 Tables). A false discovery rate (FDR)-adjusted p≤0.2 and ≤0.05 was considered significant for taxa and pathways, respectively.
Supplemental material
Supplemental material
Statistical analyses
Statistical analysis was done in GraphPad Prism V.7 (GraphPad Software, USA), STATA V.16 (StataCorp) and R V.4.2 (R Core Team, 2022). The proportions test was done to determine whether a specific variable was more frequent in different groups (eg, patients of different TB status).29 For analysis of microbiome data, non-parametric tests were used as microbiome data are not normally distributed.30 The Mann-Whitney or Wilcoxon signed rank test was used for unpaired and paired comparisons between two groups respectively (eg, α-diversity). Kruskal-Wallis with Dunn’s test was used for comparison involving more than two groups (eg, relative abundance comparisons). Spearman’s rank correlation was used to measure the association between mycobacterial relative abundance and continuous variables (eg, lymph node size). Permutational multivariate analysis of variance (PERMANOVA) was computed with 999 permutations for β-diversity differences, and R2 used to measure the proportion variation explained by a variable.20 The Benjamini-Hochberg procedure was used to correct for multiple comparisons by controlling for FDR.28 For analysis of continuous variables in different groups (eg, lymph node size different TB status), the D’Agostino-Pearson omnibus normality test was done to evaluate normality, and the relevant parametric or nonparametric test was chosen based on the normality test. A p≤0.05 was considered significant for all comparisons, unless otherwise specified.
Results
Cohort characteristics
We had 89 dTBLs, 61 nTBLs (figure 1) and 8 pTBLs (henceforth excluded due to small n), the characteristics of which are in table 1.
Demographic and clinical characteristics of patients with presumptive TBL
Lymph microbiome is distinct from background sampling controls
To assess the degree of potential carry-over from skin commensals and background DNA on equipment used for biopsies, two background DNA sampling controls were collected and subjected to the same procedures as the actual samples. We compared the microbiome in skin and saline flushes with that of the lymph fluid. Lymph fluid had similar α-diversity to background controls but different β-diversity resulting from an enrichment of Mycobacterium (online supplemental figure S1A–D), thus background contamination is unlikely.
Mycobacterium enrichment in dTBLs drives differences with nTBLs
We evaluated the overall difference between the microbial communities of TB groups by comparing their microbial diversity and composition. α-Diversity was similar in dTBLs and nTBLs (figure 2A), but β-diversity differed and Mycobacterium was the most discriminatory taxon (figure 2B,C; online supplemental figure S2 has similar comparisons with pTBLs included) appearing at several fold higher frequencies than in nTBLs (figure 2D). When patients with antibiotic use within the last year were excluded, α-diversity differences by TB status were detected (lower in dTBLs, online supplemental results). Bray distances within nTBLs were greater than within dTBLs (figure 2E), thus dTBLs were more like each other than nTBLs to each other (likely reflecting the mixture of different disease pathologies in the nTBLs and relative homogeneity of dTBLs). These results show that lymph microbial communities differ in dTBLs and nTBLs, and the microbiome of TBL is characterised by a significant enrichment of Mycobacterium.

dTBLs have a distinct microbiome to nTBLs with Mycobacterium enrichment. (A) Although α-diversity was similar, (B) β-diversity differed. Mycobacterium was enriched in dTBLs compared with nTBLs based on (C) differential abundance testing and (D) relative abundance. Discriminatory taxa appear above the threshold (red dotted line, FDR=0.2). (E) dTBLs were more compositionally similar to each other than nTBLs. dTBLs, definite-TBLs; FDR, false discovery rate; nTBLs: non-TBL; PERMANOVA, permutational multivariate analysis of variance; TBL, tuberculous lymphadenitis.
MTBC DNA found in tuberculous and nontuberculous lymph nodes
Mycobacterium reads were present in 64% (57/89) of dTBLs and 11% (7/61; p<0.0001) of nTBLs (online supplemental figure S3) and, when sequences underwent BLAST, all reads matched with Mtb, suggesting that none of these patients had environmental mycobacteria. There was a higher relative abundance of Mycobacterium reads in dTBLs (0.034 (IQR 0.001–0.460) vs 0.001 (0.001–0.001), p<0.0001; figure 2D), and the 16S rRNA gene sequencing positively correlated with TB diagnostic tests, but not with lymph node size (online supplemental figure S4A,B). These results suggest that MTBC DNA is found in most dTBL lymph nodes and occasionally occurs in nTBL lymph nodes.

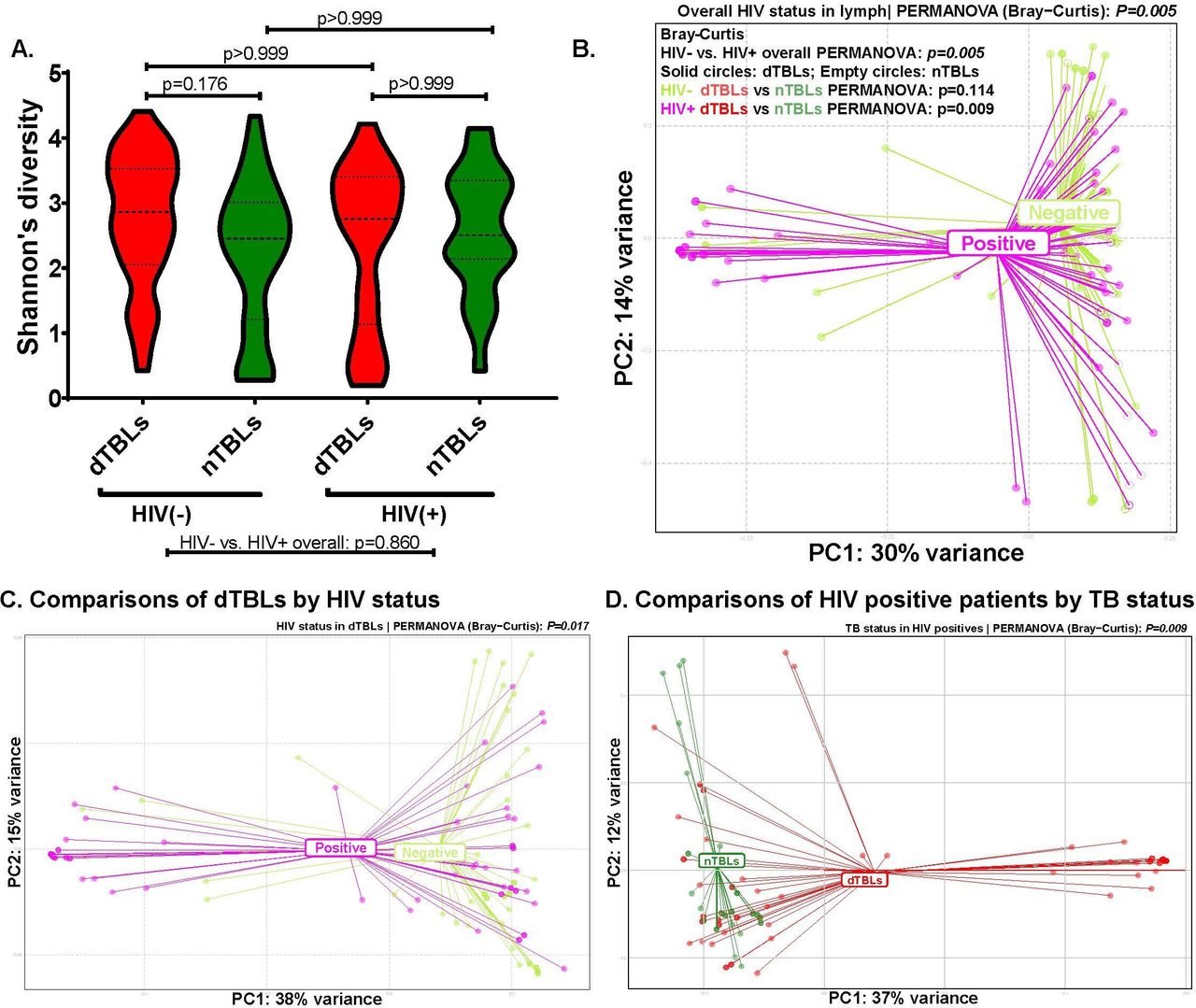
Microbiome differences in HIV-positive dTBLs versus nTBLs but not in HIV-negative dTBLs vs nTBLs. (A) α-Diversity did not differ by HIV or TBL statuses, (B) however, β-diversity differed between HIV-positives and -negatives overall (shaded circles are dTBLs, empty circles are nTBLs). β-diversity differed (C) by HIV status in dTBLs only and (D) by TBL status in HIV-positives only. d-TBLs, definite TBLs; nTBLs, non-TBLs; PERMANOVA, permutational multivariate analysis of variance; TBL, tuberculous lymphadenitis.

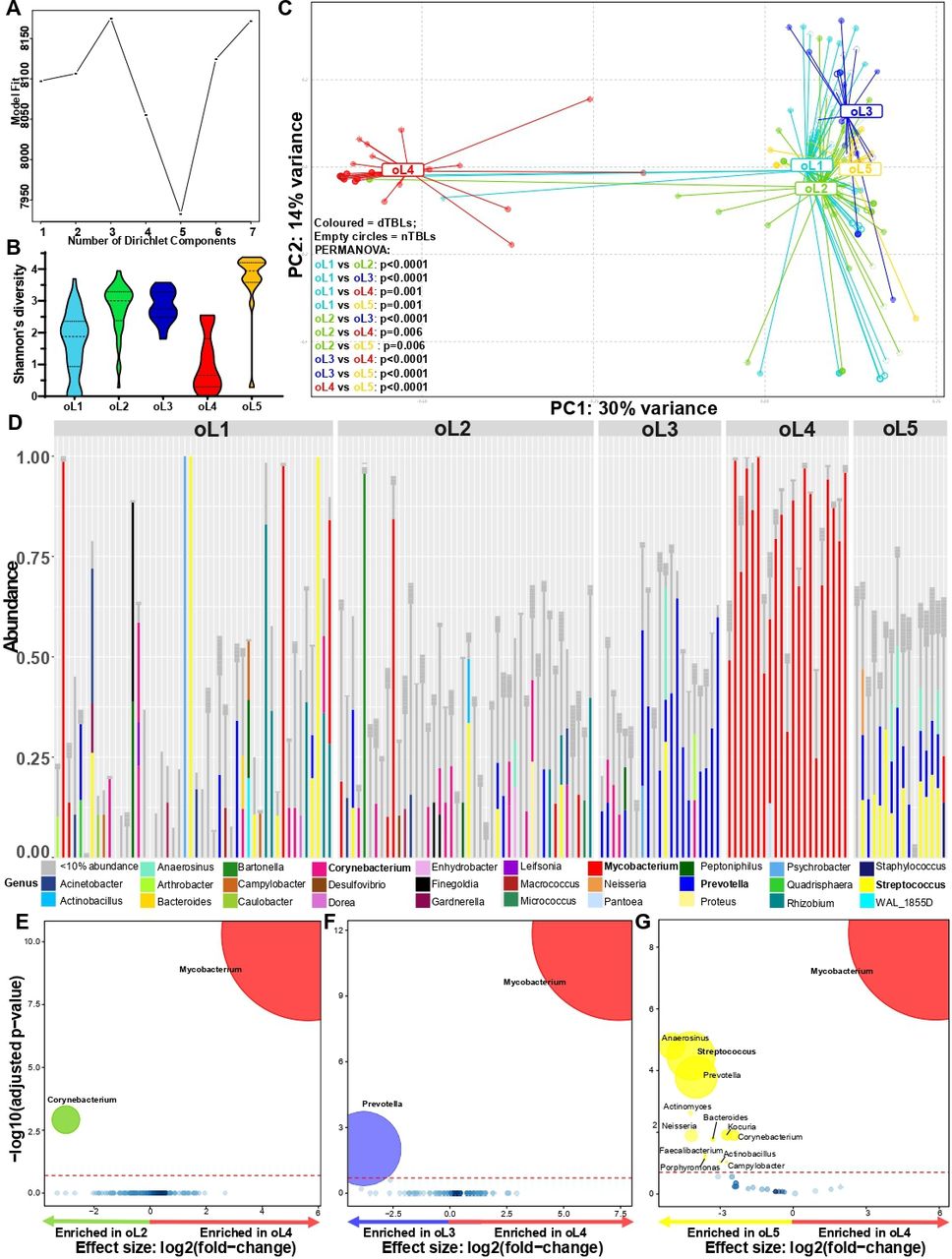
Five overall lymphotypes observed in presumptive TBL. (A) Laplace approximation identified five clusters. (B) OL5 had the highest α-diversity. (C) β-diversity differed between each lymphotype (shaded circles dTBLs, empty circles nTBLs). (D) Stacked bar plots showing OL1 with a heterogeneous mixture of genera, OL2 dominated by Corynebacterium, OL3 dominated by Prevotella, OL4 dominated by Mycobacterium, and OL4 dominated by Streptococcus. Bolded taxa represent dominating taxa. (E) Corynebacterium was enriched in OL2; (F) Prevotella enriched in oL3, (G) Mycobacterium enriched in oL4, and Streptococcus enriched in OL5. Significantly more discriminatory taxa (bolded) appear closer to the left or right and higher above the threshold (red dotted line, FDR=0.2) as significance increases. Relative taxa abundance is indicated by circle size. dTBLs, definite-TBL; FDR, false discovery rate; nTBLs, non-TBL; oL, overall lymphotype; PERMANOVA, permutational multivariate analysis of variance; TBL, tuberculous lymphadenitis.
Differences by HIV status
HIV is a known risk factor for TB. We assessed its association with the lymph microbiome first in all patients irrespective of TB status and next within dTBLs or nTBLs. Overall, α-diversity did not differ by HIV status (figure 3A), but β-diversity did (figure 3B). β-diversity differences by HIV status persisted within dTBLs (p=0.017, figure 3C) but not nTBLs. In people with the same HIV status, β-diversity differed between dTBLs vs nTBLs only in HIV-positives (p=0.009, figure 3D) where dTBLs were Mycobacterium-enriched (online supplemental figure S5B). In PERMANOVA analyses, HIV status was only significantly associated with β-diversity in dTBLs and not nTBLs (online supplemental table S2).

Three dTBL lymphotypes identified in dTBLs. (A) Best model fit based on Laplace approximation identified three clusters within dTBLs. (B) β-diversity differed between lymphotypes. (C) Stacked bar plots showing dL1 comprised of Mycobacterium and accompanying heterogenous taxa, dL2 dominated by Prevotella and Streptococus, and dL3 dominated by Mycobacterium. Bolded taxa represent dominating taxa. (D) NO taxa were enriched in dL1, (E) L2 was enriched in Streptococcus, (F) and Mycobaterium was enriched in dL3. Significantly more discriminatory taxa (bolded) appear closer to the left or right, and higher above the threshold (red dotted line, FDR=0.2) as significance increases. Relative taxa abundance is indicated by circle size. dL, dTBL lymphotype; dTBL, definite-TBL; FDR, false discovery rate; TBL, tuberculous lymphadenitis.
Lymphotype identification and their associations with clinical characteristics
We further explored this data using DMM to identify potential clusters in the TBL microbiome. These clusters were termed ‘lymphotypes’, and we evaluated associations between each lymphotype(s) and patients’ clinical characteristics.
Overall: We examined whether all patients could be grouped into distinct lymphotypes; these were termed overall lymphotypes (oLs). Five oLs with differing α-diversities and β-diversities were identified (figure 4A–C, online supplemental table S3), with the Mycobacterium-dominated (figure 4D) oL4 showing the least α-diversity. While no taxa were differentially abundant in oL1 versus other oLs (online supplemental figure S6A–C), oL2, oL3 and oL5 were enriched relative to oL4 in Corynebacterium, Prevotella and Streptococcus, respectively (figure 4E–G). The patients in all oLs were associated with distinct clinical characteristics. The majority of nTBLs occurred in highly diverse oLs with a heterogeneous mixtures of taxa; likely reflecting the spectrum of pathologies in people with TBL ruled out. oL1 was associated with characteristics indicative of less severe lymphadenitis (less TB and HIV involvement). In contrast, oL4 was associated with characteristics resembling more severe lymphadenitis (bigger lymph nodes, chylous FNABs, previous TB, HIV (with a smaller proportion of PLHIV on ART, likely to have lower CD4 counts) and TB involvement. Therefore, in summary, oL1 appears to be associated with less severe forms of lymphadenitis, whereas oL4 was associated more severe forms (online supplemental table S4).

Enriched microbial capacity for SCFA pathways in dTBLs vs nTBLs. Volcano plot depicting differentially abundant microbial pathways in dTBLs vs nTBLs inferred by PICRUSt2. key pathways of interest are bolded including aminobenzoate degradation, benzoate degradation and propanoate degradation. Significantly more discriminatory pathways appear closer to the left or right, and higher above the threshold (red dotted line, FDR=0.05) as significance increases. Relative gene abundance is indicated by circle size. dTBLs, definite-TBLs; FDR, false discovery rate; nTBLs, non-TBL; SCFA, short-chain fatty acids.
Within patients of the same TB status: We then examined whether patients within each TB group could be grouped into distinct lymphotypes. Within dTBLs, three lymphotypes (termed dTBL lymphotypes; dL) with differing β-diversities were identified (figure 5A,B), and dominated by; dL1: Prevotella and Corynebacterium; dL2: Prevotella and Streptococcus; and dL3: Mycobacterium (figure 5C–F). These dLs were termed Prevotella-Corynebacterium, Prevotella-Streptococcus and Mycobacterium, respectively. dL3s were more likely to be HIV-positive compared with dL1s, with larger lymph nodes, compared with dL1s and ddL2s. Lastly, dL2s are more likely to be female than dL1s (online supplemental table S5). Together, these differences suggest dL3 is associated with more severe TBL than other dLs. Within nTBLs, no lymphotypes were identified (online supplemental figure S7).

Predicted metagenome function reveals increased capacity for SCFA production in HIV-positive versus HIV-negative patients overall, and in dTBLs. Volcano plot depicting functional pathways differing between (A) HIV-positive and HIV-negative patients with presumptive TBL and (B) in dTBLs. Key pathways of interest include butanoate metabolism, propanoate metabolism and benzoate degradation. Significantly more discriminatory pathways appear closer to the left or right, and higher above the threshold (red dotted line, FDR=0.05). Relative pathway abundance is indicated by circle size. dTBLs, definite-TBL; FDR, false discovery rate; SCFA, short-chain fatty acids.
Predictive metagenome profiling shows increased SCFA metabolism
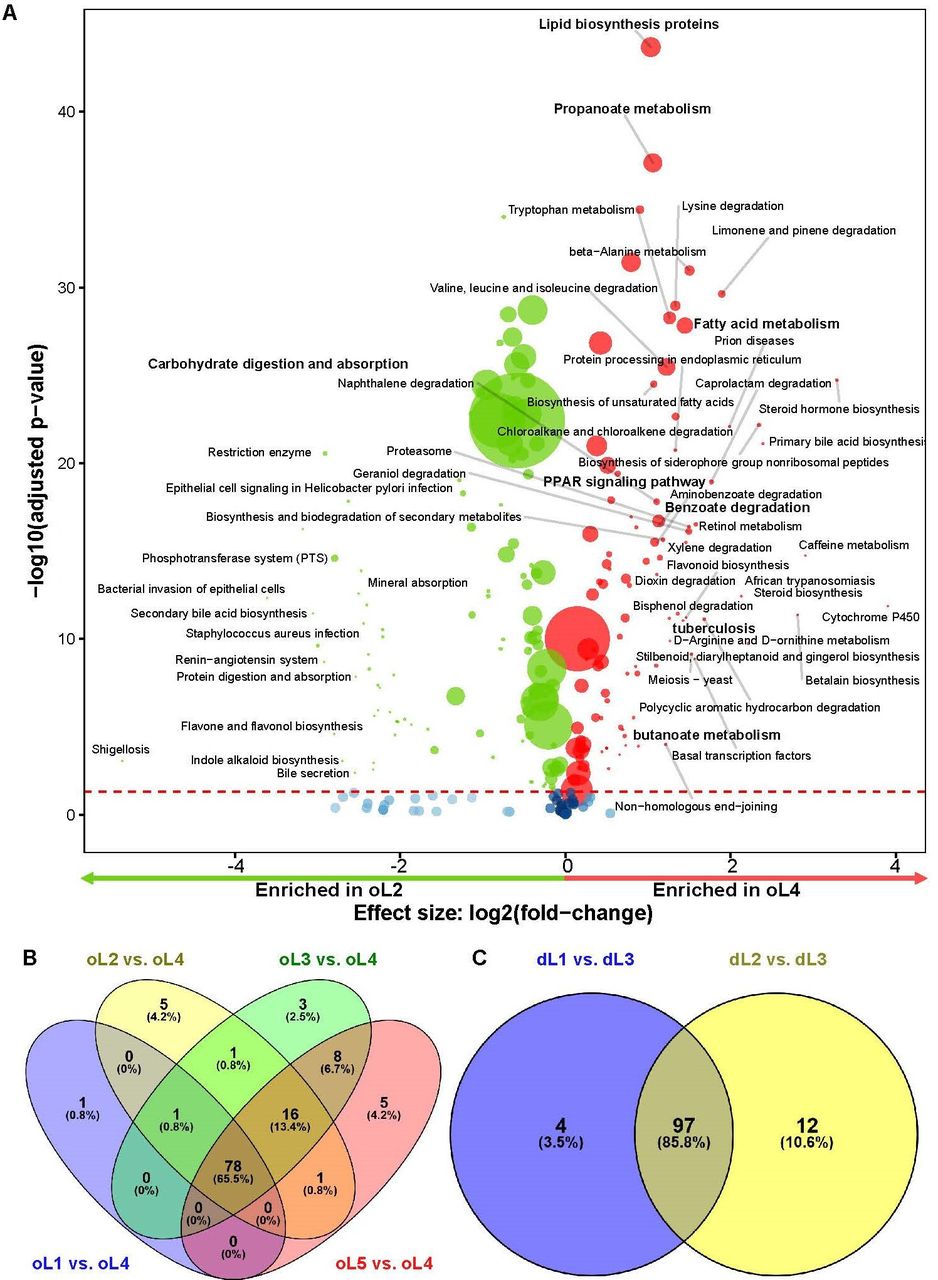
We further predicted the bacterial metagenome content and made functional inferences of the microbiome using the PICRUSt algorithm. Differences among pathways between groups were evaluated and visualised by DESeq2 analysis. In dTBLs, ‘fatty acid metabolism’, ‘benzoate degradation’, ‘propanoate metabolism’ and ‘butanoate metabolism’ were enriched, suggesting increased SCFA production (figure 6). These SCFA-related pathways were enriched in PLHIV overall and, within dTBLs (figure 7A,B).
In addition, when comparing inferred pathways in the 5 oLs, a similar core of pathways was enriched in oL4. In contrast, versus oL4, oL1 was enriched in ‘epithelial cell signalling in Helicobacter pylori infection’, oL2 and oL5 were enriched in ‘carbohydrate digestion and absorption’, and oL3 was enriched in ‘dioxin degradation’ (online supplemental figure S9A–H). When comparing the three dLs, Mycobacterium-dominated oL3 was, compared with each other dLs, enriched in the similar core pathways as the Mycobacterium-dominated oL4 in all patients (figure 8C; online supplemental figure S10). These results show that pathways involved in fatty acid-related, amino acid-related and SCFA-related inferred microbial pathways were significantly enriched in dTBLs and Mycobacterium lymphotypes (oL4 and dL3).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Differential microbial pathways between lymphotypes showing similar core pathways enriched in the Mycobacterium-dominated lymphotype. (A) Volcano plot showing differentially abundant microbial pathways inferred by PICRUSt2 in oL2 vs oL4 representing pathways enriched in oL4 compared with every other oL in all patients (overall including dTBLs and nTBLs). Significantly more discriminatory pathways appear closer to the left or right, and higher above the threshold (red dotted line, FDR=0.05) as significance increases. Relative pathway abundance is indicated by circle size. (B) 65.5% of all inferred pathways enriched in oL4 compared with each other oLS were common, while (C) 85.8% were common in dL3 compared with each other dTBL lymphotypes.Differentially enriched pathways common in all comparisons with the Mycobacterium dominant lymphotype included pathways involving lipid biosynthesis, fatty acids and SCFA metabolism, that is, lipid biosynthesis proteins, propanoate metabolism, benzoate degradation, and valine, leucine and isoleucine degradation. dL, dTBL lymphotype; dTBLs: definite-TBLs; FDR, false discovery rate; nTBLs: non-TBL; oL: overall lymphotype; SCFA: short-chain fatty acid.
Discussion
We characterised the local microbial environment in patients with lymphadenitis undergoing investigation for TB in an HIV-endemic setting. Our key findings are: (1) lymphatic microbial communities in dTBLs clustered into three distinct ‘lymphotypes’ we termed ‘Prevotella-Corynebacterium’, ‘Prevotella-Streptococcus’ and ‘Mycobacterium’, (2) the Mycobacterium dTBL lymphotype was associated with HIV-positivity and other clinical features characteristic of severe lymphadenitis and (3) dTBLs relative to nTBLs were functionally enriched in fatty acid-related, amino acid-related and SCFA-related microbial metabolic pathways with known immunomodulatory effects (the Mycobacterium lymphotype was most enriched in these pathways than other dTBL lymphotypes). Finally, (4) dTBLs without Mycobacterium reads and nTBLs with Mycobacterium reads were identified. These data show TBL at the site-of-disease is not microbially homogenous and that distinct clusters of microbial communities exist associated with different clinical characteristics. The long-term significance and importance of these lymphotypes requires prospective evaluation.
We identified three lymphotypes within dTBLs termed ‘Prevotella-Corynebacterium’, ‘Prevotella-Streptococcus’ and ‘Mycobacterium’, distinguished by different relative abundances of these taxa (Prevotella co-occurred in the first two lymphotypes). These individual taxa are enriched in respiratory secretions from pulmonary TB cases.31 32 Furthermore, within dTBLs, Streptococcus is associated with low BMI and extent of lung damage.32 Prevotella in BALF also positively correlates with SCFA concentrations and independently predicts incident TB in people without co-prevalent TB.13 Compared with the other dTBL lymphotypes, ‘Mycobacterium’ was associated with severe disease and most frequently occurred in PLHIV, agreeing with diagnostics studies that show stronger baseline mycobacterial PCR test readouts predict long term clinical outcomes in pulmonary33 and extrapulmonary TB.34 Together, these data show distinct lymphotypes are associated with different clinical characteristics and suggests that patients with the most severe Mycobacterium-dominated lymphotype may initially progress through different site-of-disease microbial states characterised by Corynebacterium, Streptococcus and/or Prevotella domination. Studies with longitudinal follow-up and repeat sampling are required to examine whether these lymphotypes have potential for clinical staging.
Importantly, Corynebacterium and Streptococcus often dominated in dTBL patients. Members of both taxa are causative agents of lymphadenitis and, even though these patients have TBL confirmed via conventional diagnostics, Corynebacterium and Streptococcus may therefore cocontribute to pathology and symptoms.35–37 Coincidently, these taxa fall within the anti-microbial spectrum of first-line TB treatment,16 meaning that this regimen may, in part, cure lymphadenitis by killing Corynebacterium and Streptococcus in addition to Mycobacterium.
Microbial pathways predicted to be most enriched in dTBLs involved fatty acid, amino acid and SCFAs (benzoate, propanoate) metabolism; all of which are associated with pulmonary TB disease compared with sick patients without TB.38 39 SCFAs in particular suppress immune pathways involved in IFN-γ and IL-17A production and, ex vivo, limit macrophage-mediated kill of Mtb. SCFA concentrations hence predict incident TB in patients.13 Our research therefore suggests that the inflammation associated with lymphadenopathy is in part caused by the presence of microbes including but not limited to Mycobacterium that are able to produce SCFAs that interfere with these immunological pathways; revealing potentially new therapeutic targets to reduce lymphadenopathy.
We detected Mtb DNA in nTBLs. These reads could be from subclinical infection, previous TB exposure or disease, where the DNA was transported to the lymph node. Mtb DNA has been found in the lymph nodes of healthy individuals and primates exposed to TB, where the sites are hypothesised to serve as a Mtb growth and persistence niche.6 dTBLs without Mycobacterium reads were also documented, however, 16S rRNA sequencing has known suboptimal sensitivity for Mycobacterium, in part due to low 16s RNA gene copy number.40
Our study has strengths and limitations. Patients were sampled once, as close as possible to treatment initiation; animal models might permit repeat invasive sampling especially if treatment is withheld. The programmatic context enabled large numbers of patients to be recruited, however, detailed long-term follow-up, which could include imaging of lymph nodes and more detailed measurements of differential responses to treatment, was not possible. We did not perform any viability tests, and since 16S gene sequencing is DNA based, the DNA may have originated from live, dead or nonculturable bacteria. Future studies could use meta-transcriptomics or culturomics to investigate this. We also used an FDR-adjusted p value threshold of 0.2 to identify differentially abundant taxa because this study was designed to be hypothesis generating and lower thresholds did not generate such taxa. Furthermore, the use of PICRUSt to infer potential function from 16S rRNA gene sequencing is a limitation. Follow-up studies using shotgun metagenomics, are necessary for inferring biological function and can more comprehensively describe the microbiota beyond bacteria. Our study was designed to describe the site-of-disease microbiome in TBL in a setting with a high burden of TB and HIV. Further research in different settings and populations is needed to validate our findings, especially those findings pertaining to microbial community clustering and the relationship between individual clusters and clinical characteristics.
In conclusion, we show dTBL patients have a distinct microbiome at the site of disease, characterised by three lymphotypes (Mycobacterium, Prevotella-Corynebacterium, Prevotella-Streptococcus). This dysbiosis of the lymphatic microbiome likely contributes to pathophysiology, including inflammatory state and clinical severity, which itself may reflect the chronicity of TB disease. TBL does therefore not appear to be a microbially homogenesis disease, and this reveals potentially new diagnosis, therapeutic and prognostic targets.
Data availability statement
Data are available on reasonable request. Data are available on reasonable request. Deidentified patient data, the study protocol, informed consent and datasets generated in this study may be requested from the corresponding author. Sequencing data reported in this paper will be released and made publicly available for sharing on publication (NCBI Bioproject PRJNA738676).
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Health Research and Ethical Committee of Stellenbosch University (N16/04/050), Tygerberg Hospital (Project ID:4134),Western Cape Department of Health (WC_2016RP15_762). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank study participants and Tygerberg Hospital FNA clinic staff especially Sr Cupido. Additionally, we thank CLIME research group staff, especially Sr Ruth Wilson and Roxanne Higgit. GRN acknowledges funding from L'Oréal-UNESCO For Women in Science Sub-Saharan Africa Young Talents Award, and the International Rising Talents Award. The content is the sole responsibility of the authors and does not necessarily represent the official views of the funders. Computations were performed using facilities provided by the University of Cape Town’s ICTS High Performance Computing team: hpc.uct.ac.za.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Presented at TBScience 2022, hosted at the Union World Conference on Lung Health 2022.
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors GRN, CCN and GT contributed to conceptualisation and design of the study and supervised the study, funding acquisition, data collection, and wrote the manuscript. GRN, CCN, IS, BW, IS, JCC, LS and GT contributed to data and statistical analysis, and figures. All authors contributed to interpretation of data and editing of the manuscript. As the study guarantor, GT is responsible for the overall content of this manuscript.
Funding This work and authors were supported by the European & Developing Countries Clinical Trials Partnership (EDCTP; project numbers SF1041, TMA2017CDF-1914-MOSAIC and TMA2019CDF-2738-ESKAPE-TB), National Research Foundation (NRF), the South African Medical Research Council (SAMRC), the Harry Crossley Foundation and Stellenbosch University Faculty of Health Sciences, and the National Institutes of Health under award numbers (R01AI136894; U01AI152087; U54EB027049; D43TW010350; K43TW012302).
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.