Article Text

Abstract
Cigarette smoking is the leading cause of preventable death worldwide. It causes chronic lung disease and predisposes individuals to acute lung injury and pulmonary infection. Alveolar macrophages are sentinel cells strategically positioned in the interface between the airway lumen and the alveolar spaces. These are the most abundant immune cells and are the first line of defence against inhaled particulates and pathogens. Recently, there has been a better understanding about the ontogeny, phenotype and function of alveolar macrophages and their role, not only in phagocytosis, but also in initiating and resolving immune response. Many of the functions of the alveolar macrophage have been shown to be dysregulated following exposure to cigarette smoke. While the mechanisms for these changes remain poorly understood, they are important in the understanding of cigarette smoking-induced lung disease. We review the mechanisms by which smoking influences alveolar macrophage: (1) recruitment, (2) phenotype, (3) immune function (bacterial killing, phagocytosis, proteinase/anti-proteinase release and reactive oxygen species production) and (4) homeostasis (surfactant/lipid processing, iron homeostasis and efferocytosis). Further understanding of the mechanisms of cigarette smoking on alveolar macrophages and other lung monocyte/macrophage populations may allow novel ways of restoring cellular function in those patients who have stopped smoking in order to reduce the risk of subsequent infection or further lung injury.
- macrophage biology
- tobacco and the lung
- respiratory infection
- emphysema
- oxidative stress
- surfactant protein
- lung proteases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- macrophage biology
- tobacco and the lung
- respiratory infection
- emphysema
- oxidative stress
- surfactant protein
- lung proteases
Introduction
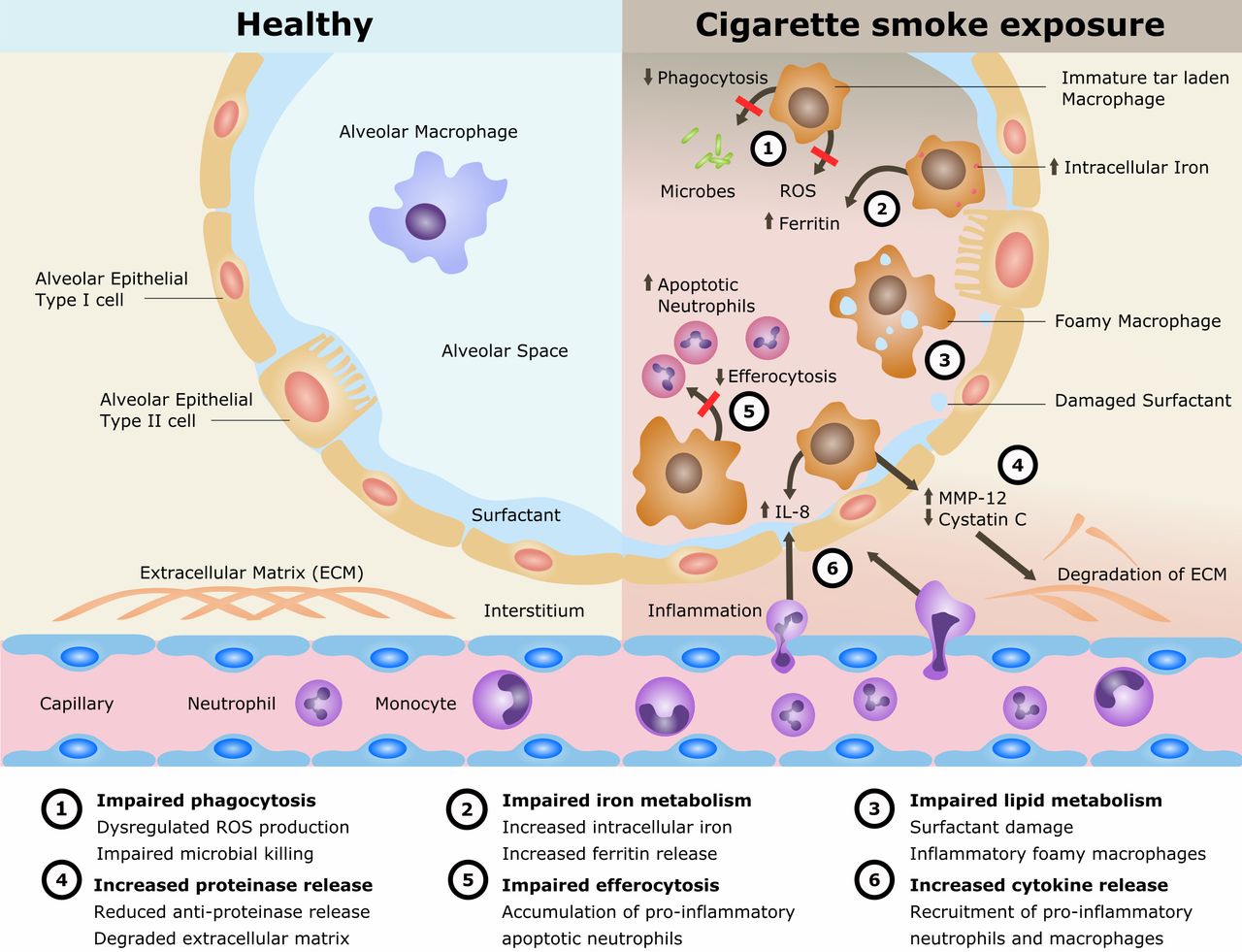
Cigarette smoking remains the single most preventable cause of death and disease worldwide and is the leading cause of chronic obstructive pulmonary disease (COPD). The disease processes in COPD are multifaceted involving oxidative stress, inflammation, proteinase/anti-proteinase imbalance, tissue destruction and inadequate repair. Alveolar macrophages (AMs) play an important role in these processes. AMs are also implicated in the cigarette smoke (CS)-related disease development of lung cancer and interstitial lung disease as well as in the increased susceptibility to pulmonary infections and acute lung injury. However, the mechanisms of CS exposure and AMs in lung disease remain poorly understood. The current understanding is reviewed (summarised in figure 1).
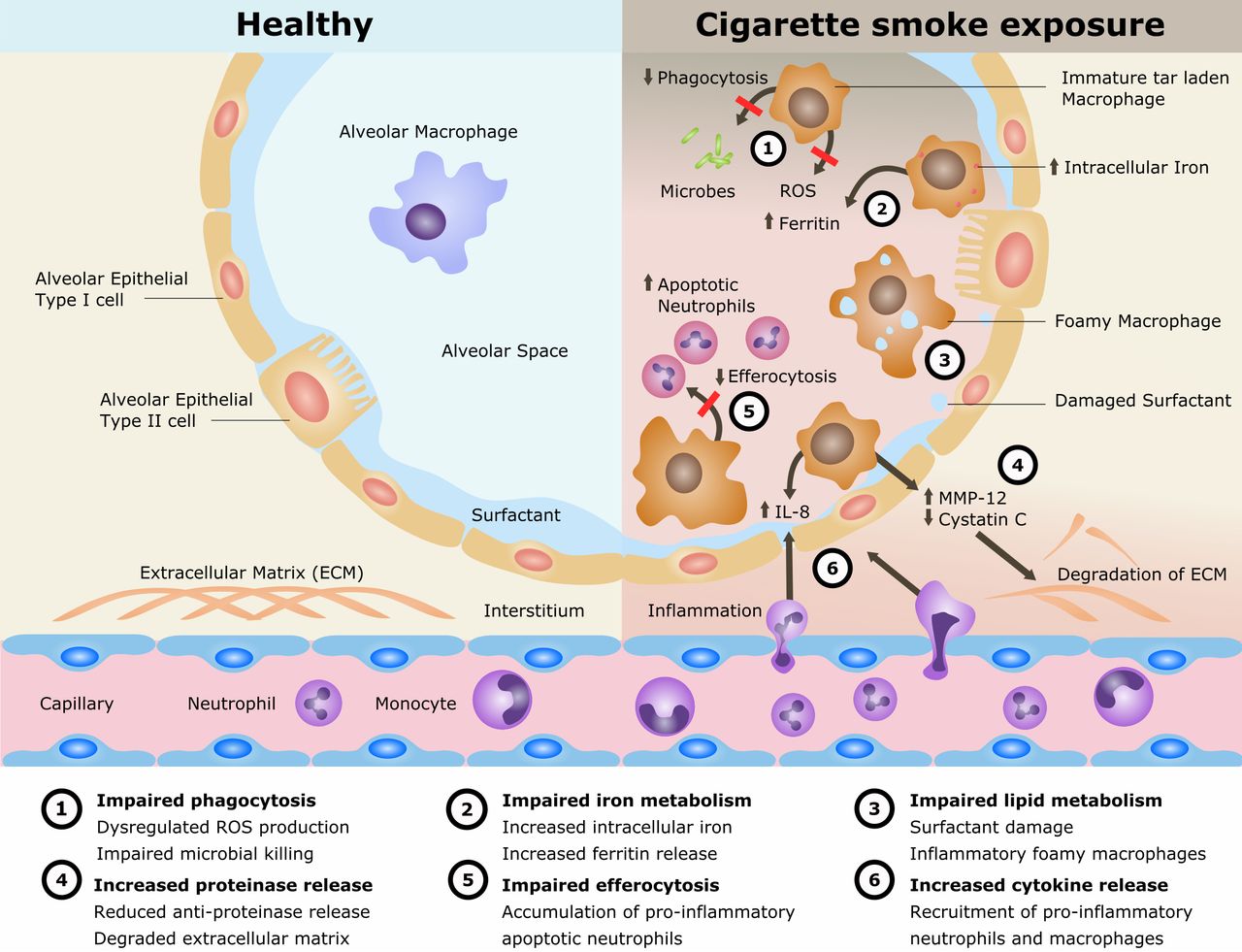
Mechanisms for disease in cigarette smoke exposure of alveolar macrophages. IL, interleukin; MMP, matrix metalloproteinase; ROS, reactive oxygen species.
Cigarette smoke
CS is an aerosol consisting of solid and liquid droplets (the particulate (‘tar’) phase) in a gaseous phase and contains over 4500 different substances,1 which have various toxic, mutagenic and carcinogenic effects. These include nicotine, tar, ammonia, carbon monoxide, carbon dioxide, formaldehyde, acrolein, acetone, polyaromatic aromatic hydrocarbons (PAHs), hydroxyquinone, nitrogen oxides and cadmium.2 Inhaled particulate matter from CS is deposited in the respiratory tract depending on the size, with larger particulates in the upper airways and smaller particulates deposited in the alveoli. CS causes oxidative stress resulting in a chronic low-grade inflammation and recruitment of inflammatory cells to the airways by activation of epithelial cells, AMs, neutrophils and T lymphocytes.3 Furthermore, CS increases the virulence of pathogens, increasing the risk of pulmonary infections.4
Alveolar macrophages
Macrophages are present in almost all tissues of the body.5 They derive their name from makros and phagein, Greek for ‘big eater’ after their primary bacterial killing mechanism, phagocytosis. In the lung, they are the most abundant immune cell present under homeostatic conditions, representing over 90% of the alveolar immune cells. As sentinel cells, AMs play an important gate-keeping role in innate immunity within the respiratory tract. AMs classically exert regulatory effects via non-specific immune-defence mechanisms such as phagocytosis, the production of inflammatory mediators such as reactive oxygen species (ROS) and the expression of inflammatory cytokines such as interleukin (IL)‐1, IL‐2, IL‐4, IL‐6, IL‐8, tumour necrosis factor‐α (TNFα) and interferon gamma (IFNγ). AMs also resolve inflammation via the release of anti-inflammatory mediators and clearance of apoptotic bodies (efferocytosis). Therefore, AMs both initiate and resolve the immune the response, as well as having a role in surfactant/lipid processing and iron homeostasis.
Ontogeny/lifespan
Once thought to originate from circulating monocytes from the bone haematopoietic stem cells (HSCs),6 mechanistic murine work has indicated that AMs are initially derived from the yolk sac in foetal development and are independent of the HSC under homeostatic conditions. It is becoming clearer that resident lung-resident AMs maintain their population during homeostasis by proliferation in situ rather than having a reliance on the macrophage precursors in the blood.7 8 Whereas during ongoing inflammation, monocytes are recruited to the lung and develop into AM-like cells.9 10 Studies looking at chimerism in lung transplant recipients have shown evidence of donor persistence of AMs,11 that can persist up to 2 years.12 However, in the inflammatory state in patients after transplant and in healthy older volunteers, the majority of human AM originate from circulating monocytes.13
Lung monocyte/macrophage populations
Distinct monocyte/macrophage populations in the lung can be defined as AMs, interstitial macrophages (IMs) and monocyte-derived cells according to their cell surface phenotype (see figure 2). Lung macrophages express the mannose receptor CD206, which has a role in recognition of microbial carbohydrates and mediating phagocytosis. This receptor is not detected in monocytes within the blood or intravascular compartment.14 AMs reside within the airway lumen and alveolar space, in contact with both the airway environment and epithelium. They are recognised by their distinctively high side scatter, high expression of CD206 and relatively low expression of monocyte marker CD14. IMs are located between alveolar epithelial cells, and unlike AMs, are negative for CD169 (Siglec-1 or sialoadhesin), a cell adhesion molecule shown to discriminate phenotype between AMs (CD206+CD169+) and IMs (CD206+CD169−).15 16 AMs can be further subdivided by levels of expression of the high affinity-scavenger receptor CD163 (CD163++ or CD163+++ subpopulations).16 Within the lung there is also a population of immature, infiltrating CD14+ pulmonary monocyte-derived cells, thought to be recently recruited into the extravascular space. These can be distinguished from the intravascular CD14+ monocytes by their expression of CD206 as well as other markers CD141, CD11c, human leukocyte antigen DR (HLA-DR) and CC-chemokine receptor type 7 (CCR7), with CD1a and CD1c allowing further categorisation of these cells.14
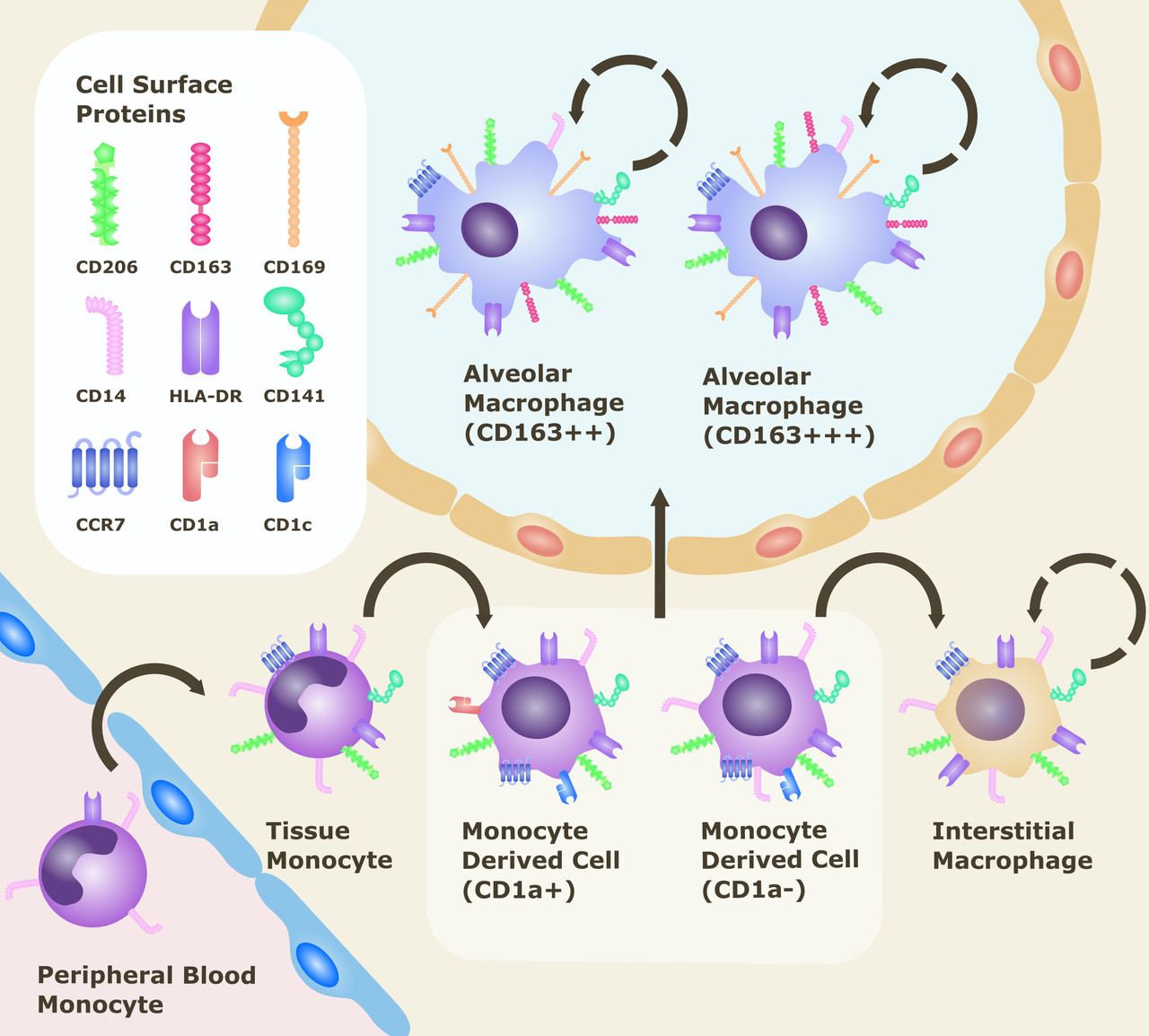
{kind=link}
{kind=link}
Cell surface markers of monocyte/macrophage populations within the lung.
Phenotype and normal function in airways
Macrophages have been shown to exhibit plasticity and can change their phenotype depending on the local environment. Macrophages have traditionally been described to form two distinct polarised phenotypes in vitro following cytokine exposure of monocyte-derived macrophages (MDMs). ‘M1 macrophages’ are classically activated, typically by lipopolysaccharide (LPS), IFNγ and/or TNFα, and are involved direct destruction of intracellular pathogens, and produce pro-inflammatory cytokines, as well as a T helper 1 (Th1)-cell environment. ‘M2 macrophages’ are alternatively activated, typically by IL-4 and IL-10 and are a more heterogeneous group and involved in down-regulating inflammation or pro-resolution and tissue repair, producing a Th2 environment. M1 and M2 have been largely unaltered since their initial classification17; M2 roles have been further categorised into M2a (parasite destruction) 2b (immune regulation) and 2c (tissue remodelling and extracellular matrix (ECM) deposition).18 With regards to cell surface markers, M1 are typically characterised by expression of HLA-DR, CD14 and CD38, while M2 are characterised by CD36, CD206 and CD163. Many studies have looked at M1/M2 with relevance to disease. However, more recently studies have moved away from this model; in vivo these phenotypes exist as part of a spectrum and many of the M1/M2 markers are co-expressed on the same macrophages, such as CD206 ubiquitously expressed by all pulmonary macrophages.14
CS exposure and AM recruitment
Increased AM and neutrophil chemotaxis
Smoking induces an increase in immune cells within the lung (4–5 fold), as assessed by bronchoalveolar lavage (BAL)19 with most of the increase attributed to the AM cellularity.20 The increased cellularity of both AM and neutrophils may be explained by chemotactic factors generated in the lung. CS exposure activates the pro-inflammatory transcription factor nuclear factor kappa B (NF-κB) and increases messenger RNA (mRNA) expression of IL-8 (one of the mediators controlled by NF-κB) within the macrophages.21 22 This process is at least partially an autologous effect, with other cells such as bronchial epithelial cells shown to release increased IL-8 in response to CS that would contribute to stimulate macrophage and neutrophil influx.23 The increased BALF cellularity may take over 1 month to significantly reduce and up to 6 months to fully normalise.24
Accumulation of autofluorescent bodies
AMs from smokers contain autofluorescent bodies25 due to the uptake of CS particles (ie, ‘tar’). Even short-term ex-vivo AM exposure to tar results in an increase in autofluorescence.26 These smoking-related inclusions can continue for up to 270 days after cessation27 and have been found on autopsy of patients who had stopped smoking 2 years prior.28 This has been shown to be the case in other studies that have investigated AMs from BAL between healthy smokers and non-smokers.25 Furthermore, the autofluorescence is not related to amount of exposure, such as pack year consumption.29 This suggests a maximum possible capacity of tar uptake, anything beyond this point will not be retained, destroying any dose response seen in earlier stage of exposure. There is also the factor of AM flux and turnover that may contribute to lack of dose effect at very high CS exposure levels.
CS exposure and AM phenotype
M1/M2 polarisation and disease states
In studies looking at the AM M1/M2 polarisation paradigm, CS has been reported to reduce phenotypic markers of M1 phenotype30 31 and/or increase markers of M2 phenotype32 33 implicated in dysregulated inflammation. Using immunohistochemistry to quantify polarisation of M1 (iNOS) and M2 (CD206) phenotype in lung tissue, in normal lungs AMs did not show M1 or M2 polarisation and instead dual expressed both markers of polarity, with the majority negative for M1 and M2 markers.30 In smokers and in COPD disease severity, AMs increased dual expression of both M1 and M2 markers.30 In early lung cancer, tumour-associated macrophages (TAMs) also showed co-expression of M1 (chemokine (C–X–C motif) ligand 9 (CXCL9)) and M2 (matrix metalloproteinase (MMP-12)) phenotypic markers,34 with single cell RNA-sequencing of TAMs showing uniform M2-like signature with varying degrees of M1-like signature. Those with >80% of TAMs co-expressing a strong (hot) M1-like signature had a stronger density of CD8+ tissue-resident memory T cells in tumours and an improved survival.
In the relationship of CS exposure and AM polarity, studies looking at gene expression profile have compared AMs to MDMs polarised to M1 or M2 in vitro using cytokines; forming either unstimulated, M1-polarised (IFNγ and LPS-treated) and M2a-polarised (IL-4-treated) macrophages.35 Microarray mRNA data suggest that smoking promotes an ‘inverse’ M1 gene expression profile defined by decreased expression of M1-induced transcripts36 and increased expression of M1-repressed transcripts with few changes in M2-regulated transcripts.37 Thus, AM M1/M2 phenotype exists as a spectrum, can be modified through CS exposure and relates to lung disease states and outcomes.
Altered cell surface marker expression and maturity
Smoking alters the phenotype of AMs, with observed differences in cell surface marker expression compared with non-smokers (table 1). Smoking increased expression of CD14 and reduced expression of CD71 in a monocyte/macrophage population from BAL.38 Absence of the transferrin receptor CD71 in AMs is characterised by reduced expression of mature markers, impaired phagocytosis and expression of profibrotic genes.39 In COPD, lung macrophages from smokers expressed lower levels of the high affinity-scavenger receptor CD163 and lower levels of the non-opsonic receptor CD36 compared with ex-smokers.40 CD163 and CD36 both have an important role in efferocytosis, which is the clearance of apoptotic cells.41 CD91 has a role in phagocytosis as part of the CD91-calrecticulin mannose receptor complex. It has been shown to be reduced in expression in AMs from healthy smokers and COPD smokers when compared with never smokers.38 CD31 (platelet endothelial cell adhesion molecule) is a surface marker that has been reported to promote tethering of apoptotic cells and is reduced in AMs from smokers.38 Blocking of CD91 and CD31 has shown to reduce AM efferocytosis.38 CD44 is involved in clearing of hyaluronan, lipid metabolism, surfactant processing and efferocytosis, all which have a role in regulating inflammation; CD44 is reduced in AM from smokers when compared with non-smokers.38 42 CD11c is an integrin that mediates cell–cell and cell to matrix interactions and is expressed at higher levels in healthy smokers compared with non-smokers.31 43 44 CD54, also known as intercellular adhesion molecule-1, has a major role in immune and inflammatory regulation and has been shown to be expressed in lower levels in AMs from smokers compared with non-smokers.43 45 CD169 is a cell marker used to characterise AMs/IMs, however, little is known about the affects of CS exposure on expression. CD169 has been shown to be involved in phagocytosis of non-typeable Haemophilus influenzae and levels reduced in COPD ex-smokers compared with non-COPD ex-smokers, however, current and recent ex-smokers (2 months) were not assessed.46
Cigarette smoking-related phenotypic changes of alveolar macrophages
Therefore, smoking influences the phenotype of pulmonary macrophages, resulting in more undifferentiated immature monocyte-like macrophages with a reduction in the mature AM surface markers needed for phagocytosis, efferocytosis, cell–cell and cell to matrix interactions. The increase in the immature macrophages seen in smokers may be due to the influx of monocyte-derived cells from the peripheral blood, with the additional increase due to AM proliferation within the lung.38
CS exposure and AM immune function
Transcriptional profile and inflammation
A number of studies have looked at the transcriptional profiles of human AMs isolated from smokers compared with non-smokers.36 47 48 The differential in gene expression belongs to the functional categories of immune/inflammatory response, lysosomal function, antioxidant-related function, signal transduction, regulation of transcription, cell adhesion, ECM and proteinase/anti-proteinase production.48 One such difference was an increase in osteopontin expression in smokers (fourfold), which was confirmed at the protein level and correlated with airflow obstruction. Osteopontin has a role in macrophage recruitment and is typically increased in response to pro-inflammatory cytokines (TNFα, IL-1β, transforming growth factor (TGF)-β).47 CS exposure has also been shown to increase TNFα, which is central to acute smoke-induced inflammation and resulting connective tissue breakdown,49 a precursor of emphysema development.
Cytochrome P450 (CYP) enzymes play an important role in activation of the CS procarcinogens to reactive metabolites that cause DNA damage,50 with CYP1B1 found to be the most highly induced gene in AMs from smokers.47 In addition, PAH–DNA adducts are higher in AMs from smokers, related to pack year consumption and were increased in smokers with higher levels of CYP3A.51 Thus, AMs play a role in the metabolism of carcinogens in CS exposure, which may contribute to lung cancer development.
Impaired bactericidal and phagocytotic processes/functions
Detection and phagocytosis of foreign particulates and microbial matter is key to AM function and, in many cases, is the first step in orchestrating an immune response to infection. More frequently, within the airway, non-opsonised phagocytosis occurs via scavenger receptors, leading to cytoskeletal rearrangements and particle engulfment.52 This process results in a phagosome, which fuses with the lysosome to form the phagolysosome, with subsequent acidification to destroy the foreign particle/organism.
Innate recognition of pathogen-associated molecular patterns (PAMPs) is mediated by evolutionarily conserved pattern recognition receptors (PRRs). Toll-like receptors (TLRs) comprise a family of PRRs that are capable of recognising distinct PAMPs. TLR2 surface marker expression is reduced in AMs from smokers and levels of mRNA and protein expression did not increase in response to LPS stimulation compared with that in non-smokers,31 suggesting that continuous exposure to LPS present in CS may down-modulate antimicrobial response.53 TLR4 surface markers also recognise LPS from gram-negative bacteria. TLR4 expression is reduced in MDMs following CS exposure,54 with associated increased intracellular ROS and reduced antioxidant glutathione, which is implicated in oxidative stress and lung inflammation and seen in the pathogenesis of emphysema.
AMs play a key role in bacterial infection. Depletion of AMs in murine models results in decreased containment and clearance of gram-negative bacteria Klebsiella pneumoniae as well as reduced overall survival.55 An in vitro study found no difference in the ability for AMs from healthy smokers to phagocytose, but a deficiency in the antimicrobial (bactericidal) properties against gram-positive bacteria Listeria monocytogenes, implying a defect in immunoregulation of the AM.56 CS extract also impaired AM phagocytosis of non-typable H. influenzae, a frequent coloniser of the upper respiratory tract, which may be due to CS potentiating reduced phosphoinositide 3-kinase signalling, triggered by the gram-negative bacteria.57 Using bioparticles from the gram-negative bacteria Escherichia coli, AM phagocytosis has been shown to be impaired in patients with COPD who were current smokers when compared with patients with COPD who were ex-smokers.40 CS exposure also causes bacteria to become more virulent, with reduced lysis and resistance to macrophage killing such as seen in gram-positive bacteria, Staphylococcus aureus.4 In this study, CS exposure altered surface charge and hydrophobicity of the bacterial cell wall, which confers resistance to killing by antimicrobial peptides, both effecting the pathogenicity as well as host susceptibility by impairment of AM function. CS also impairs AM phagocytic function against the opportunistic fungal pathogen Candida albicans in murine models after 15 min of CS exposure.58
AMs from smokers have been shown to have defective autophagy,59 a cellular function that removes unnecessary or dysfunctional cellular components and required to eliminate intracellular Mycobacterium tuberculosis.60 In addition, nicotine in CS extract has been shown to impair AM M. tuberculosis killing,61 which may play a role in the increased risk of M. tuberculosis infection in smokers.
In patients undergoing surgery, anaesthesia has also been shown to detrimentally affect AM phagocytic and antimicrobial function.62 During surgery and anaesthesia, smoking further reduces AM phagocytosis of non-opsonised and opsonised (pre-incubated with rabbit immunoglobulin G anti-bovine serum albumin)62 albumin-coated fluorescent particles and killing of L. monocytogenes, which were halved compared with non-smokers.63 The expression of pro-inflammatory cytokines (TNFα, IFNγ, IL-1β) was increased only half as much in smokers.63 This supports the observed increased risk in postoperative pulmonary complications in patients who smoke before surgery. The effect of smoking cessation has been investigated64; 4 hours after induction of anaesthesia, the decreases in antimicrobial function against L. monocytogenes were 1.5–3 times greater in AMs from current and recent ex-smokers (2 months) compared with never smokers. AMs from current and mid-term ex-smokers (2–6 months) mounted a blunted inflammatory response with only a 50%–20% increased expression of pro-inflammatory (TNFα, IFNγ, IL-1β) and anti-inflammatory (IL-10, IL-4) cytokines compared with never smokers.
Proteinase/anti-proteinase imbalance and tissue destruction
One of the causes of smoking-related lung damage is through the imbalance of proteolytic enzymes (proteinases) and their inhibitors secreted by AMs. MicroRNA (miRNA) profiling studies in human AMs found that the most strongly downregulated miRNA in smokers compared with non-smokers was miR-452,37 which has a role in inhibiting the expression of MMP-12, a proteinase which degrades elastin and causes emphysema.65 The same study found that global miRNA expression was 50%–60% less in AMs from smokers.37 This may be explained by the reduced expression of the cytosolic RNA endonuclease (DICER),66 that has a key role in cleaving precursor miRNAs into mature functional 20–22-nucleotide miRNAs. MMP-12 has been shown to be the third most highly induced gene in smokers (ninefold) compared with non-smokers47; MMP-12+ macrophages are also increased in BAL in smokers and those with patients with COPD.67 Furthermore MMP-12 can increase the expression of placenta growth factor (PGF), upregulating downstream signalling molecules of PGF and resulting in bronchial epithelial cell apoptosis and emphysema.68 AMs also produce elastolytic cysteine proteinases that have the capacity to cause significant lung destruction, particularly in an acidic environment.69 Cathepsin S is another potent elastase, with expression and activity found to be increased in current smokers.70 Cystatin C (CysC) is a major constitutive secretory product of AMs and is the most important inhibitor to cysteine proteinases which are also produced by the cells. CysC forms complexes with cathepsins and regulates proteinase secretion or leakage from dying or diseased cells. CysC release has been shown to be downregulated in response to CS.71 Thus, CS tips the balance of proteinase/anti-proteinase release from AMs, contributing to cell death alongside inadequate repair, with elastolysis and connective tissue destruction, all of which are involved in the pathology of emphysema.
Dysregulated ROS production
ROS is essential for innate immune response of AMs and is produced by the enzyme NADPH Oxidase 2 (NOX2). Activation of NOX2 requires translocation of cytoplasmic subunits (p40 phox , p47 phox and p67 phox ) and the guanosine triphosphate-binding Ras-related C3 botulinum toxin substrate 2 (Rac2) to a membrane-bound heterodimer cytochrome.72 CS has shown to increase intracellular ROS and activate the cellular energy sensor AMP-activated protein kinase in a nicotine dependent manner, resulting in increased IL-8 cytokine production,73 which plays a role in lung inflammation.
Cadmium is a heavy metal of considerable toxicity found in CS, which has been shown to influence the phagocytic and microbiocidal capacity of murine AMs.74 Cadmium levels were significantly elevated (ninefold) in BAL from human smokers, which correlated with loss of membrane Rac2 and p67 phox localisation and NOX2-derived ROS synthesis.75 In the same study, in CS exposed mice, cadmium inhibited Rac2 activation needed for the generation of ROS in lung macrophages, recovery of Rac2 function reduced Streptococcus pneumoniae bacterial burden and increased survival. This may explain, in part, a mechanism for lower respiratory tract infection susceptibility in smokers.
Mitochondrial dysfunction and oxidative stress
Mitochondria are essential organelles that act as the powerhouse of the cell by generating high amounts of energy though ATP by oxidative phosphorylation. There is increasing interest in the alteration of mitochondrial activity as a mechanism for AM dysregulation in CS-related lung disease. AMs from patients with COPD had increased basal mitochondrial ROS (mROS) expression, though were unable to increase mROS production in S. pneumoniae infection, thus reducing the late phase of intracellular bacterial killing.76 MDMs from COPD increased mROS in response to oxidative stress, but with a decreased mitochondrial membrane potential, indicating dysregulated mitochondrial function.77 The differences in these studies may reflect that AMs from the lungs are primed to have elevated mROS following bacterial exposure, whereas MDMs are yet to be primed to reveal the intrinsic defect described. Further understanding of the role of CS in mitochondrial function in AMs is needed as a potential therapeutic approach for improving phagocytosis and infective exacerbations in COPD.
CS exposure and AM homeostasis
Surfactant/lipid processing and inflammation
The AM has a role in regulating the surfactant layer coating the alveolar epithelium by clearing damaged and oxidised surfactant produced by the alveolar epithelial type II (AT2) cell ensuring low surface tension to prevent alveolar collapse.78 AMs contribute to around 50% of surfactant turnover through degradation, with the remaining surfactant uptake and recycling by AT2 cells.79 Granulocyte macrophage colony-stimulating factor (GM-CSF) is required for the differentiation and functional activity of the AM in mice80 and has an important role in surfactant homeostasis. Absence of GM-CSF in humans is seen in pulmonary alveolar proteinosis, in which AMs are unable to catabolise surfactant lipids and proteins, resulting in an accumulation of pulmonary surfactant-associated proteins in the airways and associated inflammation.
CS exposure is associated with damaged pulmonary surfactant, inflammation and dysfunctional processing and lipid accumulation in AMs (a defining feature of foam cells); IL-1α-dependent inflammation following exposure is required to maintain surfactant homeostasis in the lungs through production of GM-CSF.81 In another murine study, CS exposure increased pulmonary GM-CSF and AM accumulation. Chronic GM-CSF exposure induced features of diffuse interstitial pneumonia, a smoking-associated parenchymal lung disease associated with AM accumulation, increased MMP-12 secretion, parenchymal lung disease and emphysema.82
Reverse lipid transport is crucial in pulmonary homeostasis and allows AMs to properly export intracellular lipids and cholesterols, most of which originate from the pulmonary surfactant. ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) have important roles in the exporting cell, as they bind to apolipoprotein A-1 (ApoA-1), the principal component of high-density lipoproteins (HDLs). Deficiencies of ABCA1 and ABCG1 increase lipid accumulation in AMs and chronic lung inflammation in mice.83 84 CS affects pulmonary expression of ABCA1 and ABCG1 in both humans and mice, with ApoA-1 deficient mice showing a 50% reduction in reverse lipid transport capacity, and increased lung neutrophilia and larger macrophage size in response to CS exposure.85 There is a potential role for augmenting reverse lipid transport. ApoA-1 overexpression in mice had a protective effect in attenuating inflammation and development emphysema following CS exposure.86
Iron homeostasis and oxidative damage
Iron homeostasis is important in the lung, as excess iron, particularly in the ‘free’ form produces toxic reactive hydroxyl radicals, and favours intracellular bacterial growth such as in M. tuberculosis infection.87 Intracellular iron is increased in AM from smokers88 89 and in patients with COPD and lung cancer.89 Release of extracellular iron and ferritin is higher in AMs from smokers compared with non-smokers and more-so in heavy smokers.90 CS and crocidolite (blue asbestos) exposure have been shown to have a synergistic role in promoting extracellular ferritin release by AMs.91 Thus, the increased extracellular iron from AMs is a potential source of oxidative damage and inflammation in the lung following CS exposure.
Efferocytosis and resolution of inflammation
Efferocytosis is the clearance of primarily apoptotic neutrophils in the setting of acute lung inflammation. During efferocytosis the cell membrane of the AM engulfs the apoptotic cell, forming a vesicle called the efferosome, preventing the apoptotic cell from breaching its membrane and leaking toxic elements such as enzymes, oxidants and proteinases into the surrounding tissue. Thus, efferocytosis requires the AM to adopt an anti-inflammatory state in order to prevent inflammatory responses to self-proteins.92
Efferocytosis is reduced in AMs from smokers compared with never-smokers.93 AMs recognise apoptotic cells using cell surface receptors including CD44 (hyaluronan receptor), which is expressed at lower levels in smokers38 and is important for clearance of apoptotic neutrophils and the release of anti-inflammatory and pro-repair mediators such as TGF-β.94 CS has been shown to reduce efferocytosis in AMs through the interaction of AM with CS modified ECM proteins and aberrant surfactant processing.95 Defective efferocytosis of AMs may also be driven by the significantly increased levels of CS-induced oxidative stress.96 Another mechanism for reduced efferocytosis may be due to CS-induced defects in phagosome and the lysosome fusion, demonstrated in rat models.97
CS-reduced AM efferocytosis was restored through the delivery of GM-CSF to the alveolar space and was associated with reduced morbidity following influenza infection.98 In patients with COPD, AM efferocytosis was higher than in those who stopped smoking compared with those who continued to smoke, suggesting a CS-related effect on AM in COPD that may be partially resolved on smoking cessation.38 Following community acquired pneumonia, smokers had reduced AM efferocytosis, as were those who were not on a statin; suggesting that smoking and statins may have antagonistic effects on the Rac1 and RhoA pathway in influencing efferocytosis and inflammation resolution.99 CS is known to affect pathways leading to the activation and membrane localisation of the enzyme Rac, which facilitates the cytoskeletal rearrangements needed for efferocytosis.100 Statins are a potential therapy to improve AM efferocytosis, as demonstrated in AMs from mice and patients with COPD.101 Macrolides including azithromycin are another potential therapy to rescue defective efferocytosis as shown in patients with COPD,102 possibly due to an upregulation of CD206 but the mechanism remains to be fully elucidated. Glucocorticoids improve AM efferocytosis but decrease phagocytosis in murine models of S. pneumoniae.103 Thus, resolving efferocytosis may leave the AMs that have ingested apoptotic cells less able to recognise and kill bacteria.104 Therefore a balance must be struck between microbial clearance and pro-resolution.
Conclusion
CS has a plethora of effects on AMs with changes in phenotype, phagocytosis and bacterial killing, ROS production, proteinase/anti-proteinase release, iron and lipid homeostasis and efferocytosis. Subsequent acute and chronic inflammation with inflammatory cell recruitment and resultant destruction/remodelling increases susceptibility to pulmonary infection and development of CS-induced lung diseases. Many of these smoking-induced changes on the AM persist following cessation in smoking, with the duration of cessation needed for partial or full resolution of phenotype and function largely unknown. E-cigarette vaping has shown similar effects to smoking on AM function,105 with shared mechanisms to CS exposure, though the perception is that they are a safer alternative. Further understanding of the mechanisms of CS on AMs and other lung monocyte/macrophages populations may allow novel ways of restoring cellular function in those patients to reduce risk of infection or further lung injury.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors DRT and BN conceived the review. STL, AS and DP drafted the final manuscript. All authors have critically appraised and approved the final version for submission.
Funding AS and DRT are funded by the Medical Research Council (MR/L002736/1).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.