Article Text

Abstract
Background Conflicting evidence has emerged regarding the relevance of smoking on risk of COVID-19 and its severity.
Methods We undertook large-scale observational and Mendelian randomisation (MR) analyses using UK Biobank. Most recent smoking status was determined from primary care records (70.8%) and UK Biobank questionnaire data (29.2%). COVID-19 outcomes were derived from Public Health England SARS-CoV-2 testing data, hospital admissions data, and death certificates (until 18 August 2020). Logistic regression was used to estimate associations between smoking status and confirmed SARS-CoV-2 infection, COVID-19-related hospitalisation, and COVID-19-related death. Inverse variance-weighted MR analyses using established genetic instruments for smoking initiation and smoking heaviness were undertaken (reported per SD increase).
Results There were 421 469 eligible participants, 1649 confirmed infections, 968 COVID-19-related hospitalisations and 444 COVID-19-related deaths. Compared with never-smokers, current smokers had higher risks of hospitalisation (OR 1.80, 95% CI 1.26 to 2.29) and mortality (smoking 1–9/day: OR 2.14, 95% CI 0.87 to 5.24; 10–19/day: OR 5.91, 95% CI 3.66 to 9.54; 20+/day: OR 6.11, 95% CI 3.59 to 10.42). In MR analyses of 281 105 White British participants, genetically predicted propensity to initiate smoking was associated with higher risks of infection (OR 1.45, 95% CI 1.10 to 1.91) and hospitalisation (OR 1.60, 95% CI 1.13 to 2.27). Genetically predicted higher number of cigarettes smoked per day was associated with higher risks of all outcomes (infection OR 2.51, 95% CI 1.20 to 5.24; hospitalisation OR 5.08, 95% CI 2.04 to 12.66; and death OR 10.02, 95% CI 2.53 to 39.72).
Interpretation Congruent results from two analytical approaches support a causal effect of smoking on risk of severe COVID-19.
- COVID-19
- clinical epidemiology
- tobacco control
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. The individual participant data from UK Biobank is available on application by bona fide researchers to the UK Biobank (via the Access Management Team). Individual level data is not permitted to be shared by the authorship team. A data dictionary defining ICD-10 codes used to define comorbidities, or Read/SNOMED codes to define smoking exposures from primary care data, can be made available on request to the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Does cigarette smoking increase risk of severe COVID-19?
What is the bottom line?
In this study using UK Biobank, we obtained congruent results from observational analyses (n=421 469) and Mendelian randomisation analyses (n=281 105) regarding increased risk of COVID-19-related hospitalisation and death in smokers.
Why read on?
Together, the results from our two analytical approaches support a causal effect of smoking on the risk of severe COVID-19.
Introduction
Observational evidence accruing throughout the COVID-19 pandemic has identified several factors associated with COVID-19 severity, including older age, male sex, cardiometabolic comorbidities (eg, hypertension and diabetes) and non-white ethnicity.1–3 However, evidence on the role of smoking in COVID-19 has been inconsistent.4–9 Several studies conducted early in the pandemic reported a lower prevalence of active smokers among COVID-19 patients relative to the general population, and a large population-based study conducted in the UK found that smoking was associated with lower risks of COVID-19 mortality10 on adjustment for multiple prognostic factors. In contrast, current smoking was associated with higher risks of COVID-related death, adjusted for age and sex, in another large population-based study (OpenSAFELY),2 higher risks of self-reported SARS-CoV-2 infection in online surveys,11 an increased burden of COVID-19 symptoms in the ZOE COVID-19 symptom study12 and increased risk of severe COVID-19 with respiratory failure in a Mendelian randomisation study of lifetime smoking.13 Furthermore, several large-scale meta-analyses have concluded that a history of smoking is associated with a range of adverse outcomes including severe COVID-19 and mortality.5 6 ,8 Alongside the conflicting observational evidence, the commencement of clinical trials of nicotine therapy, based on the hypothesis that nicotine could inhibit penetration and propagation of SARS-CoV-2 (eg, NCT04598594,14 NCT0458341015) further emphasise the need for greater clarity on the relationship between smoking and COVID-19 to ensure appropriately informed public health messaging.
Various limitations of observational studies of COVID-19 have been proposed. The majority of such studies undertaken in the early stages of the pandemic were conducted primarily in hospitalised patients and/or in communities with limited SARS-CoV-2 testing, prompting concerns that observed associations between smoking and COVID-19 could be distorted due to selection bias,16 17 with potential for so-called ‘collider’ bias of particular relevance.17 18 In the context of COVID-19, hospitalisation/testing may lead to collider bias if both smoking and COVID-19 increase the likelihood that an individual will be tested or hospitalised. Consequently, studies conducted within tested or hospitalised patients may yield biased estimates of smoking and SARS-CoV-2 infection and any downstream consequences of infection (ie, death). Moreover, conventional observational studies are subject to limitations such as residual confounding and/or reverse causation, inappropriate adjustment (eg, if adjustment variables lie on causal pathways) and remain focused on association rather than causation. Mendelian randomisation (MR) can overcome some of these limitations by using genetic variants as proxies for smoking behaviours and thereby provide genetic support for causal associations. Thus, alongside robust observational analyses, MR analyses can provide complementary insights to enhance our ability to assess potentially causal relationships.
In UK Biobank, we investigated associations between smoking and COVID-19 using both observational and Mendelian randomisation approaches. First, we performed multivariable regression to assess associations of smoking behaviours with SARS-CoV-2 infection, COVID-19-related hospitalisation, and COVID-19-related death among 421 469 participants. Second, we used two-sample MR to examine the causal relevance of smoking initiation (ie, propensity to begin smoking) and smoking heaviness for COVID-19 outcomes in 281 105 White British participants.
Methods
Study design, data sources and participants
UK Biobank recruited over 500 000 individuals aged 40–69 years across the UK between 2006 and 2010.19 Participants were genotyped and underwent a comprehensive baseline assessment with biofluid collection. In addition, approximately 4–5 years after the baseline visit, a subset of the cohort (~20 000 individuals) underwent a repeat assessment of the baseline measures (ie, ‘resurvey’).
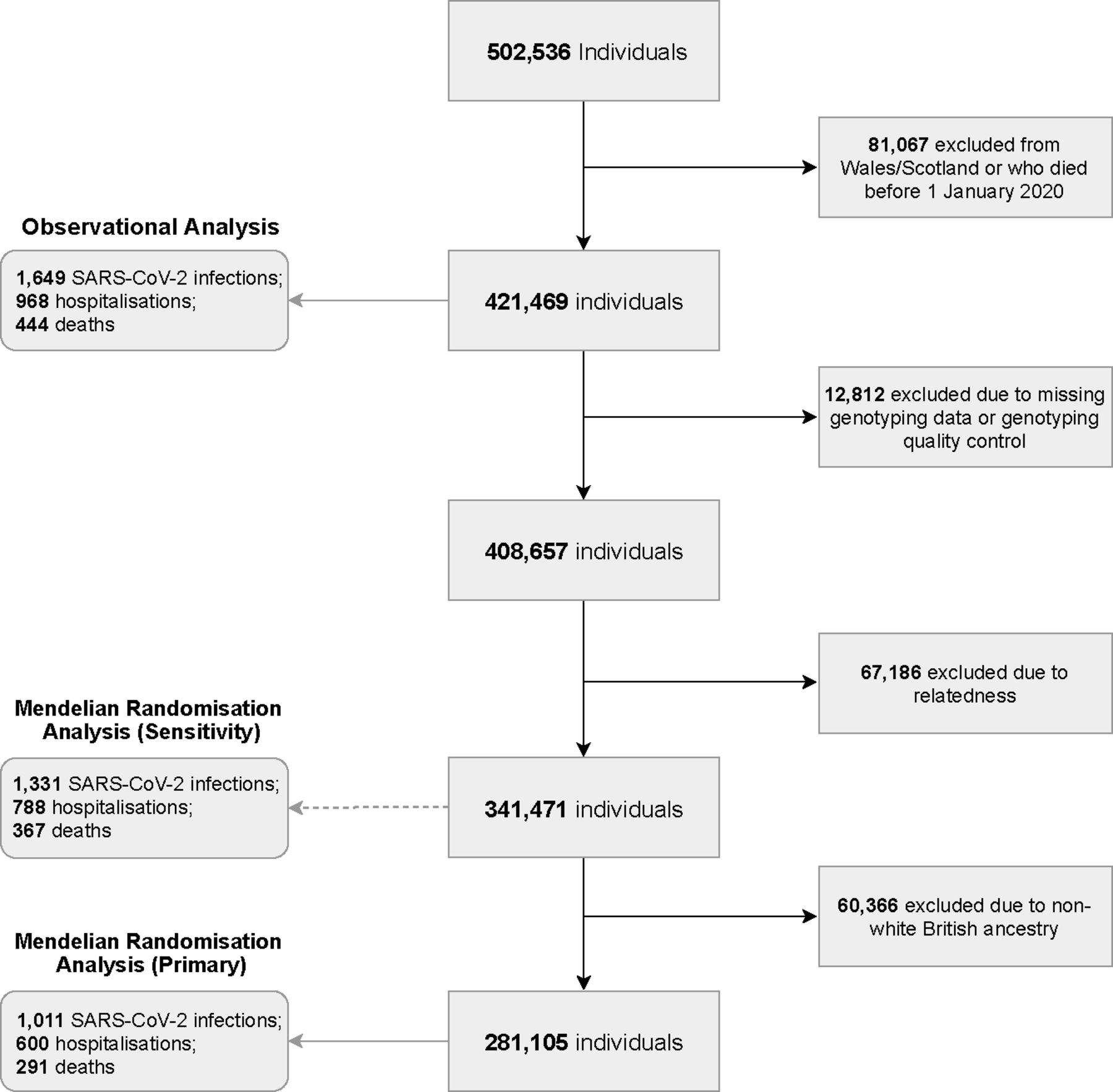
In the present study, UK Biobank participants that were alive on 1 January 2020 (consistent with the emergence of COVID-19 diagnoses), and who were resident in England (as linkage to SARS-CoV-2 testing data was limited to England) were considered. Dynamic linkage to Public Health England’s Second Generation Surveillance System microbiology database enabled ascertainment of positive cases of SARS-CoV-2.20 Primary care data were available via linkage to EMIS and TPP electronic healthcare record software systems. Linked Hospital Episode Statistics (HES) provided data on hospital admissions across National Health Service (NHS) commissioning groups in England. Lastly, national death registry linkage was used to ascertain COVID-19-related deaths. Exposure and outcome data available up to 18 August 2020 were included. Figure 1 demonstrates the derivation of the study cohorts for both the observational and MR analyses, based on common inclusion criteria and distinct exclusion criteria where relevant.
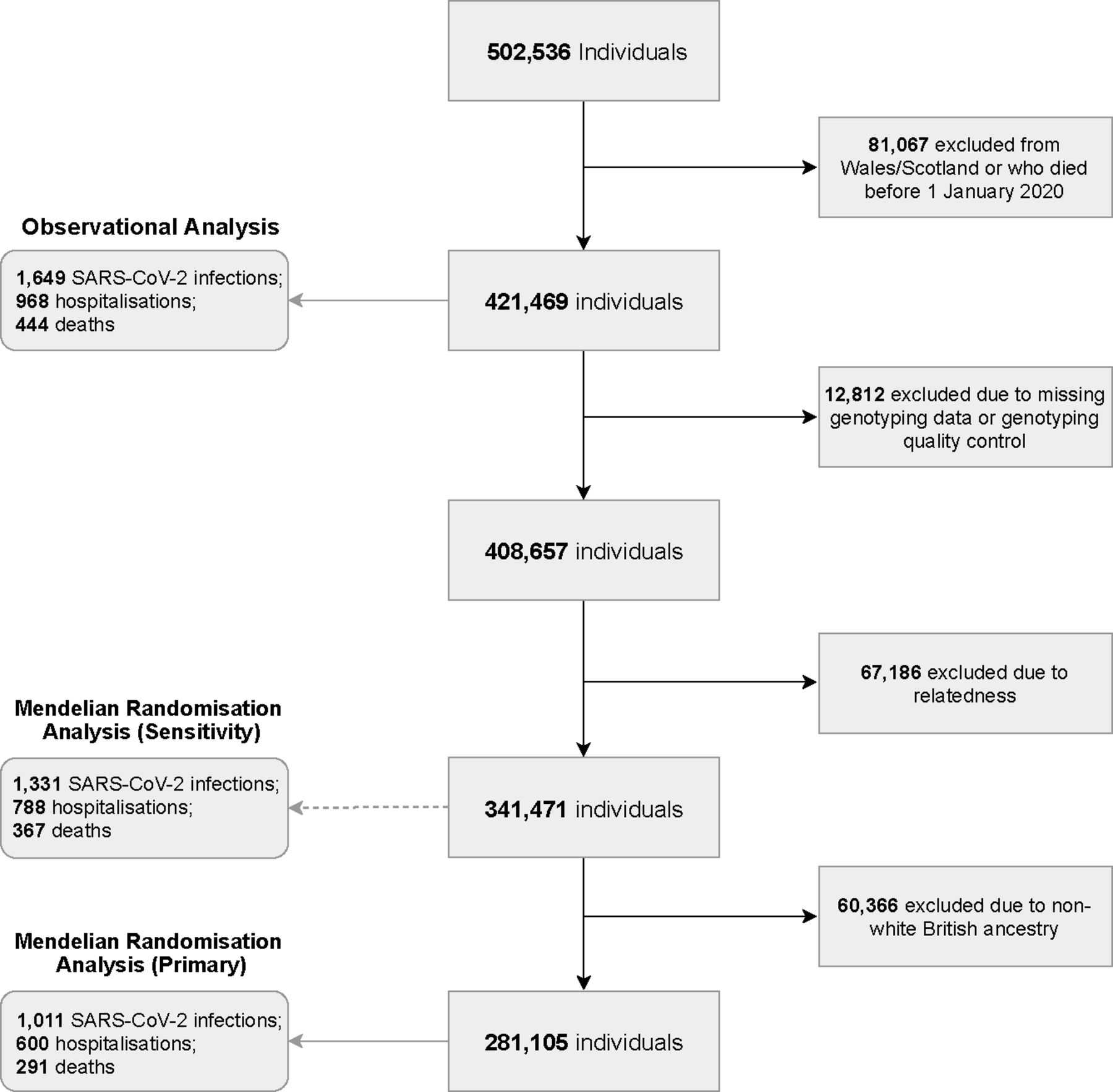
Flow chart of study derivation using UK Biobank.
COVID-19 outcomes
Study outcomes were confirmed SARS-CoV-2 infection, COVID-19-related hospitalisation, and COVID-19-related death. Confirmed SARS-CoV-2 infection was defined as a positive RT-PCR test result on the Public Health England microbiology database. COVID-19-related hospitalisation was defined as a confirmed or suspected COVID-19 diagnosis (ICD10 codes U07.1, U07.2) on a hospital admission record and was not conditional on a positive test. COVID-19-related death was defined as the presence of COVID-19 as a primary or contributory cause of death based on mortality register data (ICD10 codes U07.1 or U07.2) and was not conditional on a positive test or hospitalisation. In each analysis, cases were defined as participants who experienced the COVID-19-related outcome of interest; controls were participants without a record of any COVID-19-related outcome.
Smoking exposure variables for observational analyses
For observational analyses, we classified smoking in two ways. First, participants were classed as never-smokers, former smokers and current smokers. Thereafter, for the main observational analysis, participants were classified into five groups in terms of smoking heaviness: never-smokers, former smokers, light smokers (<10 cigarettes/day), moderate smokers (10–19 cigarettes/day) and heavy smokers (≥20 cigarettes/day). This was based on the most recently recorded smoking status (prior to 1 January 2020) available for each individual either from UK Biobank baseline or resurvey assessments (from touchscreen assessment data) or linked primary care records. The most recent recorded smoking status was taken as the exposure.
Touchscreen questionnaires completed at the UK Biobank baseline assessment were used to define participant age at study origin (40–49, 50–59, 60–69, 70–79 and 80+ years), sex, ethnicity (eight categories), Townsend deprivation quintile, body mass index (kg/m2) (underweight (<18.5), healthy weight (18.5–24.9), overweight (25–29.9), obese (30–34.9) and severely obese (>35)). Self-reported non-cancer illness codes recorded at the nurse-led interview during recruitment into UK Biobank were used to derive smoking- and non-smoking related comorbidities at baseline (see table 1 for full list).
Demographic and clinical characteristics of the observational study cohort from UK Biobank
Genetically predicted smoking behaviours for Mendelian randomisation analyses
For the MR analyses, we generated genetic proxies for smoking initiation and for smoking heaviness based on genome-wide significant (p<5×10-8) genetic variants identified in published meta-analyses of genome-wide association studies (GWAS and Sequencing Consortium of Alcohol and Nicotine Use; GSCAN).21 GSCAN was conducted in individuals of European ancestry and included 24 studies of smoking initiation (n=1 232 091) and 25 studies of smoking heaviness (n=337 334).
Genetic instruments were constructed using genetic variants identified as conditionally independent using the partial correlation-based score statistic method in the published GSCAN analyses.21 22 The resulting smoking initiation genetic instrument included 378 conditionally independent genetic variants associated with smoking initiation, defined as a binary phenotype based on either having smoked >100 cigarettes over an individual’s life course, smoked every day for at least a month, or ever smoked regularly. The smoking heaviness genetic instrument included 55 conditionally independent variants associated with smoking heaviness, which was defined as the average number of cigarettes smoked per day. All MR analyses are reported per SD higher phenotype. For smoking initiation, the SD represented the weighted average prevalence determined in the GSCAN meta-analysis.21
Statistical methods
Observational analyses
Multivariable logistic regression was used to estimate odds ratios (ORs) and 95% confidence intervals (CIs) for the effects of smoking exposures on COVID-19 outcomes. Our model adjustment strategy was guided by directed acyclic graphs to identify potential causal pathways between smoking and COVID-19 outcomes (online supplemental figure 1). For each outcome of interest, the association with smoking was serially adjusted for: (1) age and sex; (2) age, sex, ethnicity, deprivation quintile and non-smoking-related comorbidities (interstitial lung disease, cystic fibrosis, bronchiectasis, chronic liver disease and diabetes (type 1 or 2)) to assess the ‘total effect’ and (3) age, sex, ethnicity, deprivation quintile, non-smoking-related comorbidities, smoking-related comorbidities (lung cancer, asthma, chronic obstructive pulmonary disease, hypertension, ischaemic heart disease, congestive cardiac failure, chronic kidney disease and atrial fibrillation) and body mass index to assess the ‘direct effect’. To assess the utility of more contemporaneous exposure ascertainment via primary care, we repeated these analyses using the baseline-derived UK Biobank smoking status.
Supplemental material
As a confirmed infection is conditional on receiving a COVID-19 test in the context of highly selective sampling during the earlier stages of the pandemic,16 we also performed a sensitivity analysis using inverse probability weighting23 for the confirmed infection endpoint to adjust for each participant’s likelihood of receiving a COVID-19 test. Inverse probability weights were calculated using a logistic regression model incorporating the confounders included in the relevant model. Estimates for COVID-19 hospitalisation and death did not undergo such sensitivity analyses as their ascertainment was not dependent on receiving a positive test.
All observational analyses were undertaken as complete case analyses due to limited data missingness (<1.8%). We computed the ‘E-value’ for ORs to assess the robustness of observed associations to unmeasured confounding.24 The E-value is defined as the minimum strength of association, on the risk ratio scale, that an unmeasured confounder would need to have with the exposure and outcome to explain away an observed exposure-outcome association (conditional on the measured covariates).24
Mendelian randomisation analyses
For MR analyses, of the 421 469 included in the observational analyses, 12 812 were excluded due to missing genotype data or failure of UK Biobank conducted quality control,25 and a further 67 186 due to relatedness (third degree or closer). Primary MR analyses were restricted to 281 105 participants of White British ancestry, in accordance with the population in which the genetic instruments were derived and to limit the potential impact of population stratification (see figure 1). Sensitivity analyses including 341 471 participants, unrestricted by ancestry, were also conducted.
Single nucleotide polymorphism (SNP)-outcome associations were generated for each genetic variant in the instruments using logistic regression with adjustments for gender, genotype array, and the first 10 principal components of population structure. For the smoking heaviness analyses, associations with individual genetic variants were calculated in those who had ever smoked. Mean F-statistics were calculated to assess the strength of the genetic instruments; values >10 indicate adequate instrument strength.26 The random-effects inverse-variance weighted (IVW) method27 was used to estimate causal effects of smoking behaviours on COVID-19-related outcomes, with estimates shown as ORs and 95% CIs per SD difference in the genetically proxied smoking behaviour. Specifically, causal effects are reported per 1 SD difference in genetically determined prevalence of smoking initiation and cigarettes smoked per day for the two instruments, respectively. For smoking initiation, the SD represented the weighted average prevalence determined in the GSCAN meta-analysis.21 Weights were based on the SNP-exposure estimates from GSCAN as described above. The potential bias in the weights arising from participant overlap between GSCAN and UK Biobank (31% for smoking initiation and 36% for smoking heaviness) was examined in sensitivity analyses in which we repeated primary analyses using SNP-exposure estimates that excluded UK Biobank and 23andMe participants (as weights excluding UK Biobank alone were not publicly available).
The IVW method provides causal effect estimates with optimal precision but assumes that all genetic variants included are valid instrumental variables. To examine the robustness of the MR estimates to departures from this assumption, we conducted methodologic sensitivity analyses, which included the weighted median method28 (in which up to half of the genetic variants are permitted to be invalid instrumental variables); the Mendelian randomisation-Egger (MR-Egger) method29 (in which all genetic variants are permitted to be invalid instrumental variables, provided that the pleiotropic effects are independent of instrument strength) and the Mendelian randomisation-Pleiotropy Residual Sum and Outlier (MR-PRESSO) method,30 which performs a pleiotropy residual sum and outlier test and allows detection and correction of pleiotropy via outlier removal. Heterogeneity between variants that may indicate pleiotropy was investigated using Cochran’s Q (IVW) and Rucker’s Q (MR-Egger) statistics, and the MR-PRESSO pleiotropy residual sum and outlier test. The validity of the MR-Egger method was evaluated using the regression dilution I2(GX) statistic.31 If the result was <0.9, then SIMEX corrections were performed. Finally, we performed Steiger directionality tests to determine if the observed effects were directionally causal.32
Analyses were performed in R v4.0.2 using the MendelianRandomization,33 TwoSampleMR34 and MRPRESSO30 packages.
Results
Observational associations between smoking behaviour and COVID-19 outcomes
We identified 421 469 individuals eligible for inclusion in observational analyses (figure 1). These participants had a median age of 68.6 years (IQR 60.6 to 73.7) and the majority were female (55.1%) and of white ethnicity (93.5%). Demographic and clinical information for these study participants is shown in table 1. During the study period, 13 446 (3.2%) individuals underwent a SARS-CoV-2 test, and 1649 (0.4%) had a PCR-confirmed infection. Some 968 (0.2%) had a COVID-19-related hospitalisation (of whom 154 (15.9%) were known to have been admitted to critical care) and 444 (0.1%) had a COVID-19-related death. Of the 444 deaths, 188 (42.3%) had no record of COVID-19-related hospitalisation; and of 968 hospitalised individuals, 256 (26.4%) died of COVID-19.
The most recent smoking status recorded prior to the start of the study (1 January 2020) was derived from UK Biobank questionnaires for 122 296 people (median time between recorded status and study origin 10.6 years (IQR 9.9 to 11.3); TPP for 70 333 people (median time 4.5 years, IQR 0.9 to 13.1) and EMIS for 225 628 people (median time 1.1 years, IQR 0.4 to 3.4). Overall, the smoking status used in the analyses, which included 13 711 current smokers (3.3%), represented behaviours current as of an average of 2.3 years (IQR 0.5 to 9.5 years) prior to the start of the pandemic. Table 2 summarises the concordance between UK Biobank and the most recently recorded smoking status; online supplemental tables 1 and 2 summarise concordance between UK Biobank and individual primary care databases.
Concordance of smoking status as per latest UK Biobank record and from latest of any of UK Biobank or linked primary care datasets (ie, the smoking exposure used in the final analyses)
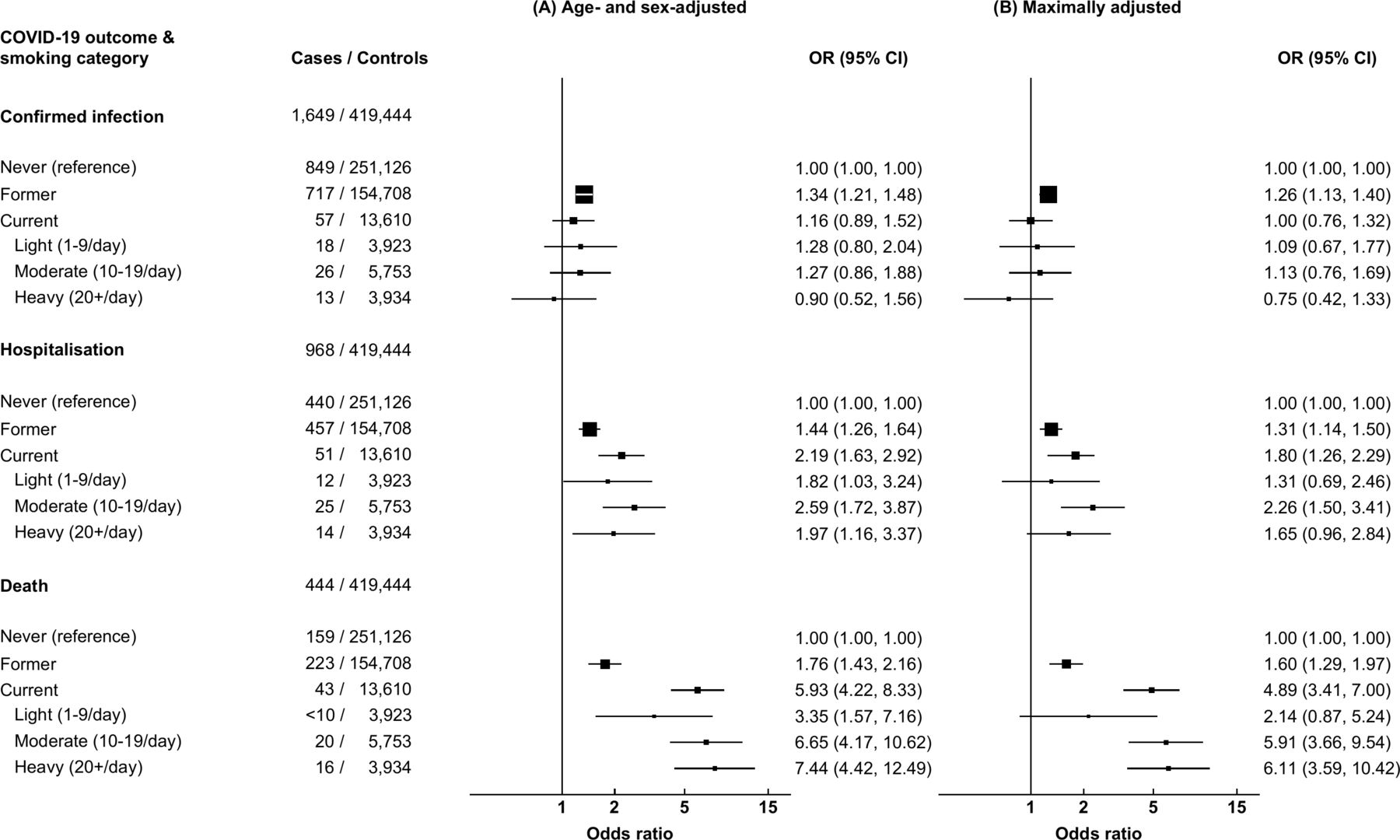
Of those receiving a SARS-CoV-2 test, 7071 (52.6%) were never-smokers, 5684 (42.3%) were former smokers, 136 (1.0%) smoked 1–9 cigarettes/day, 240 (1.8%) smoked 10–19 cigarettes/day and 191 (0.9%) smoked 10+ cigarettes/day. Compared with never-smokers, former smokers had a higher risk of confirmed infection on adjustment for age and sex (OR 1.34, 95% CI 1.21 to 1.48) and on maximal adjustment (OR 1.26, 95% CI 1.13 to 1.40; figure 2). There was no evidence that current smoking conferred a higher risk of infection relative to never smoking, either on adjustment for age and sex (OR 1.16, 95% CI 0.89 to 1.52) or on full adjustment (OR 1.00, 95% CI 0.76 to 1.32). When weighting by the likelihood of having received a SARS-CoV-2 test (online supplemental table 3), heavy smoking (≥20 cigarettes/day) was associated with a reduced risk of confirmed infection when adjusting for age and sex (OR 0.50, 95% CI 0.29 to 0.89) and on maximal adjustment (OR 0.55, 95% CI 0.31 to 0.99; online supplemental table 4).
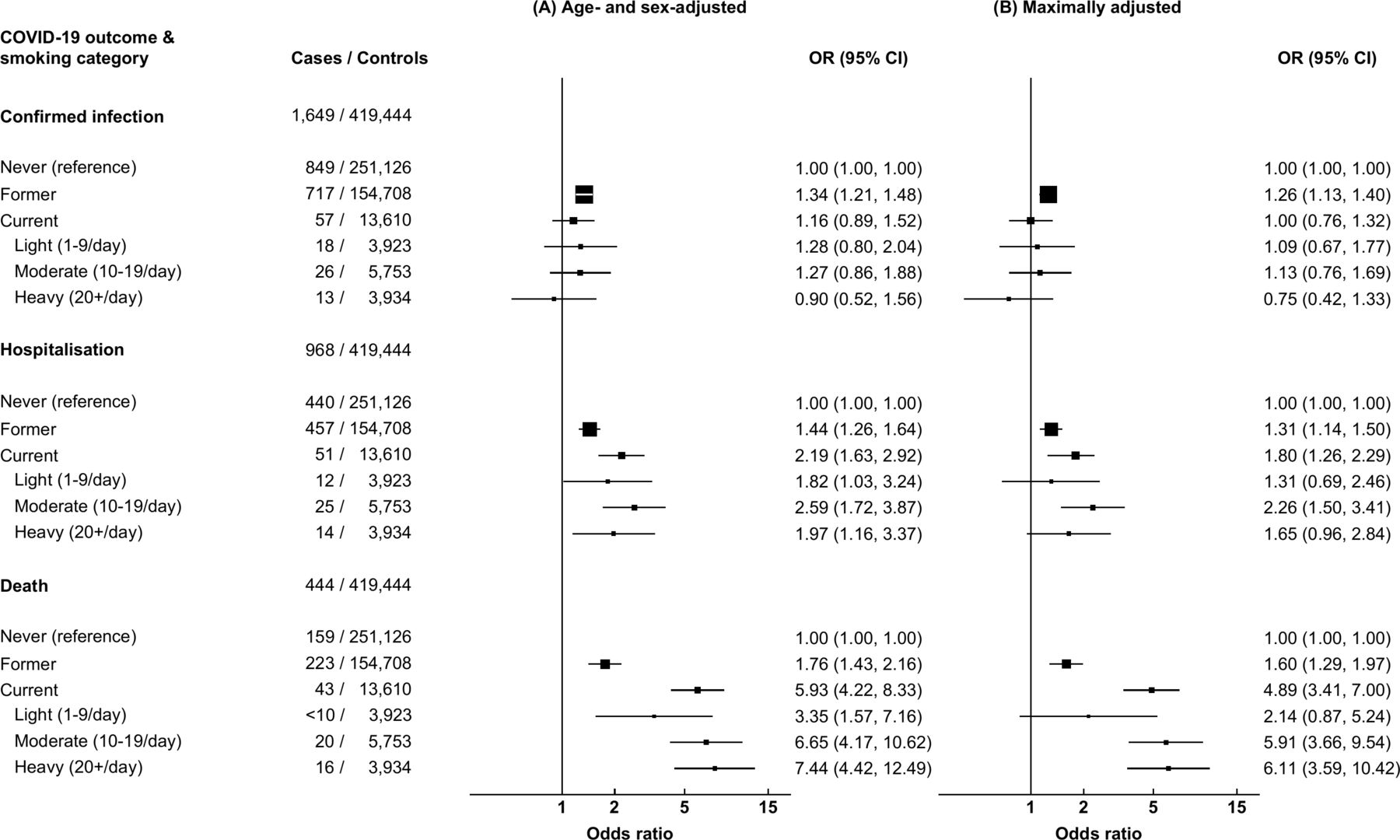
Results from multivariable logistic regression models examining the effect of observed smoking behaviours on COVID-19 outcomes with serial adjustment guided by directed acyclic graphs. CI, confidence interval; COVID-19, novel coronavirus disease 2019; OR, odds ratio. (A) Model adjusted for age and sex. (B) Model adjusted for age, sex, ethnicity, deprivation, interstitial lung disease, cystic fibrosis, bronchiectasis, chronic liver disease, diabetes, lung cancer, asthma, chronic obstructive pulmonary disease, hypertension, ischaemic heart disease, congestive cardiac failure, chronic kidney disease, atrial fibrillation and body mass index. Controls are individuals that did not experience any of the outcomes of interest, that is, positive SARS-CoV-2 RT-PCR test, hospital admission for confirmed or suspected COVID-19, or died due to confirmed or suspected COVID-19.
Former smoking and current smoking were associated with higher risks of COVID-19-related hospitalisation on adjustment for age and sex (OR 1.44, 95% CI 1.26 to 1.64; OR 2.19, 95% CI 1.63 to 2.92, respectively) and on maximal adjustment (OR 1.31, 95% CI 1.14 to 1.50; OR 1.80, 95% CI 1.26 to 2.29, respectively). There was a consistent, positive association between smoking and risk of COVID-19-related death. For heavy smokers, the ORs for COVID-19-related death compared with never-smokers were 7.44 (95% CI 4.42 to 12.49) when adjusting for age and sex, and 6.11 (95% CI 3.59 to 10.42) in the fully adjusted model.
In the sensitivity analysis using only baseline UK Biobank-derived smoking status, similar relationships were observed, although the ORs were attenuated (online supplemental table 5).
Mendelian randomisation analyses
MR analyses included up to 281 105 individuals (restricted to unrelated White British participants; figure 1). Analyses of genetically predicted smoking initiation included 1011 cases of confirmed infection (0.4%), 600 COVID-19-related hospitalisations (0.2%) and 291 COVID-19-related deaths (0.1%). Among 114 080 ever-smokers included in the analysis of smoking heaviness, there were 503 confirmed infections (0.4%), 328 COVID-19-related hospitalisations (0.3%) and 180 COVID-19-related deaths (0.2%).
Association of genetic variants with smoking behaviours in UK Biobank
To illustrate the strength of the genetic instruments, associations within UK Biobank were assessed. Genetic variants predicting lifelong differences in smoking initiation (mean F statistic 44.96) and smoking heaviness (mean F statistic 85.91) were strongly associated with observed smoking behaviours. A 1 SD difference in the weighted genetic instrument for smoking initiation was associated with higher odds of having ever smoked (OR 2.73, 95% CI 2.64 to 2.81, p<1.0×10-300) and a 1 SD difference in the genetic instrument for smoking heaviness was associated with 6.6 higher cigarettes smoked per day among ever smokers (n=75 846) (coefficient 6.62, 95% CI 6.69 to 7.12, p=7.1×10-206).
Genetically predicted smoking initiation on COVID-19 outcomes
A 1 SD higher genetically predicted propensity to initiate smoking was associated with 45% higher odds of confirmed SARS-CoV-2 infection (ORIVW 1.45, 95% CI 1.10 to 1.91, p=0.01; figure 3). Comparable causal estimates were observed for COVID-19-related hospitalisation (ORIVW 1.60, 95% CI 1.13 to 2.27, p=0.01) and COVID-19-related death (ORIVW 1.35, 95% CI 0.82 to 2.22, p=0.23), although the association with death was not statistically significant.

{kind=link}
{kind=link}
{kind=link}
Results from Mendelian randomisation analyses (inverse-variance weighted estimates) examining the effects of genetically predicted smoking behaviours on COVID-19 outcomes. CI, confidence interval; COVID-19, novel coronavirus disease 2019; OR, odds ratio; SD, standard deviation.
In methodological sensitivity analyses, the causal estimates were largely consistent (online supplemental table 6). However, the MR-Egger estimate for COVID-19-related death was directionally discordant with a wide confidence interval (OREgger 0.35, 95% CI 0.04 to 2.82, p=0.33). Across all three COVID-19 outcomes, tests for heterogeneity showed no evidence of horizontal pleiotropy and the MR-Egger intercept suggested that the IVW results gave a consistent estimate of the causal effect (online supplemental table 7). Steiger directionality tests supported a conclusion of valid causal effects, and all I2(GX) values supported validity of the estimates under the no measurement error assumption (online supplemental table 7). Results of comparable direction and magnitude were obtained using SNP-exposure estimates that excluded UK Biobank (online supplemental table 8), suggesting inferences were insensitive to potential bias arising from participant overlap between GSCAN and UK Biobank.
Genetically predicted smoking heaviness and COVID-19 outcomes
Genetically predicted smoking heaviness was associated with 2.5-fold higher odds of confirmed SARS-CoV-2 infection (ORIVW 2.51, 95% CI 1.20 to 5.24, p=0.01 per SD higher number of cigarettes smoked per day; figure 3). Furthermore, the causal estimates were notably stronger for more severe COVID-19 phenotypes, including COVID-19-related hospitalisation (ORIVW 5.09, 95% CI 2.04 to 12.67, p<0.001) and COVID-19-related death (ORIVW 10.02, 95% CI 2.53 to 39.73, p=0.001).
In methodological sensitivity analyses, the effects were directionally consistent across different MR methods, and showed no evidence of heterogeneity or directional pleiotropy (online supplemental table 6). Results of comparable direction and magnitude were obtained using SNP-exposure estimates (ie, weights) that excluded UK Biobank, though the magnitude of effects was slightly attenuated (online supplemental table 8).
Sensitivity analyses among all 341 471 unrelated participants, independent of ancestry, were comparable, although slightly attenuated relative to the primary analyses for both smoking instruments considered (online supplemental table 9).
Discussion
This is the first study to combine conventional observational analyses and MR to investigate the association between smoking and COVID-19, and results from both approaches were congruent regarding increased risks of COVID-19-related outcomes being associated with smoking. The E-values were 3.0 (lower CI 1.8) for hospitalisation and 9.3 (lower CI 6.3) for death, suggesting it is unlikely that unmeasured confounding could nullify the observed associations between smoking and severe COVID-19. Results from MR analyses showed that genetic variants predicting lifelong differences in smoking initiation and smoking heaviness were associated with higher risks of SARS-CoV-2 infection as well as severe COVID-19-related outcomes, providing genetic support for a causal relationship between these smoking behaviours and the COVID-19 phenotypes examined. Our results complement and extend those from survey-based studies demonstrating increased odds of COVID-19 symptoms and symptomatic burden in current smokers,11 12 as well as MR analyses observing an association between lifetime smoking and the risk of hospitalisation and respiratory failure due to COVID-19.13
Observational meta-analyses of mostly hospitalised patients have been undertaken with divergent results and are of varying quality.4 6 7 Two recent population-level studies in the UK have reported seemingly protective effects of cigarette smoking on COVID-19 severity, either as parameters in a single, mutually adjusted regression model,2 or specified explicitly as a confounder for the effect of ACE inhibitors.10 However, the system of covariates used in their regression models differed, complicating comparisons of mutually adjusted HRs, and such estimates can themselves be problematic in light of the ‘table 2 fallacy’.35 Notably, a harmful effect of smoking was seen on adjustment for age and sex in one of these studies.2 Aside from the degree of selection into study cohorts, contemporaneity of exposures is a key consideration when analysing exposure-outcome associations. For example, a recent analysis of UK Biobank, which also showed an increased risk of severe COVID-19, used smoking status on enrolment.9 Our study sought to triangulate observational and MR evidence, and was able to address several limitations of other studies, such as by utilising linked UK Biobank and primary care data, and minimising collider bias by avoiding conditioning analyses on positive test results.
Our MR results recapitulate and expand on a previous investigation of the role of smoking and other cardiometabolic traits in the development of sepsis and severe COVID-19.13 Using outcome data from the COVID-19 Host Genetics Initiative, investigators found that a genetically predicted lifetime smoking index (a genetic composite of smoking initiation, duration and heaviness) was associated with increased risk of severe COVID-19 with respiratory failure and hospitalisation. Our study extends this work by demonstrating causal relationships between smoking and multiple COVID-19 phenotypes, including SARS-CoV-2 infection and death, while also disentangling the effects of multiple smoking exposures on these traits.
Our study had several strengths and potential limitations. Strengths of the observational analysis included the use of both UK Biobank-collected data from initial recruitment and data ascertained from primary care records to identify the most recent smoking status prior to the pandemic (at an average of 2.3 years previous). The discordance between UK Biobank-derived smoking status and the most recent available smoking status (driven predominantly by primary care data) varied by smoking heaviness, and the magnitude of effect estimates were smaller using UK Biobank baseline data from a median of 10.6 years prior to the pandemic. This suggests that as smoking behaviours change over time, smoking status collected at initial UK Biobank assessment does not adequately capture these behaviours at the start of the COVID-19 pandemic. This emphasises the value in use of primary care and other longitudinal records linked to UK Biobank participants when examining dynamic phenotypes in emerging diseases, or conditions in which temporality of exposure is relevant. UK Biobank also has a selection bias towards healthy volunteers,36 37 so observational results from the current study, which included older participants (mean age 69 years) with lower rates of smoking than the UK population as whole (3.3% vs 14.1%), may not necessarily generalise to the wider UK population. Self-reported smoking status may also be unreliable and risk misclassification of true exposure, but more objective measures such as cotinine levels were not available.
A strength of the MR framework is that results are less susceptible to environmental confounding and reverse causation, and we performed a range of analyses to assess the robustness of our findings to violations of instrumental variable assumptions. One limitation is that the smoking GWAS was conducted in White British individuals, which could limit generalisability to other populations. Second, there is evidence that genetically predicted effects on smoking initiation may be partially mediated by impulsivity-related traits (eg, risk-taking) that influence the decision to initiate smoking.38 Consequently, it is possible that the relationship between genetically predicted smoking initiation and COVID-19 could represent a propensity to engage in risk-taking behaviours that increase risk of infection, such as refusing to wear a mask or to appropriately socially distance. However, the additional finding of an association between genetically predicted smoking heaviness and COVID-19 outcomes lends further support to our findings.
A limitation of both the observational and MR approaches is the selective SARS-CoV-2 testing during the present study, which may have resulted in ascertainment of infections that were less representative (eg, more symptomatic cases) of infections generally. During the study period, testing was concentrated in the hospital setting and therefore many infections, such as those in frail care home residents where risk of severe disease is highest but hospital admission avoidance is common, were never confirmed on RT-PCR. Our study therefore sought to combine suspected and confirmed COVID-19 for our endpoints of interest – while we are unable to confirm the true number of actual infections, we believe this was a less biased approach than only analysing confirmed cases.
Overall, the congruence of observational analyses indicating associations with recent smoking behaviours and MR analyses indicating associations with lifelong predisposition to smoking and smoking heaviness support a causal effect of smoking on COVID-19 severity.
Supplemental material
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. The individual participant data from UK Biobank is available on application by bona fide researchers to the UK Biobank (via the Access Management Team). Individual level data is not permitted to be shared by the authorship team. A data dictionary defining ICD-10 codes used to define comorbidities, or Read/SNOMED codes to define smoking exposures from primary care data, can be made available on request to the corresponding author.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AKC and AvE are joint first authors.
JH-C and JCH are joint senior authors.
Twitter @AshClift
Contributors AKC: conceptualisation, study design, data analysis, writing first draft of manuscript. AvE: conceptualisation, study design, data analysis, writing first draft of manuscript. PST: conceptualisation, study design, data analysis, revision of manuscript. HMS: conceptualisation, study design, revision of manuscript. NL: conceptualisation, study design, revision of manuscript. CACC: study design, advice on data analysis, interpretation, revision of manuscript. MRM: conceptualisation, study design, revision of manuscript. PA: conceptualisation, study design, revision of manuscript. JH-C: conceptualisation, study design, revision of manuscript. JCH: conceptualisation, study design, data analysis, revision of manuscript. AKC, PST and JH-C have verified the underlying observational data, and AvE and JCH have verified the underlying genetic data.
Funding AKC is supported by a Clinical Research Training Fellowship from Cancer Research UK (DCS-CRUK-CRTF20-AC). HMS and MRM work in a research unit funded by the UK Medical Research Council (MC_UU_00011/7). HMS is also supported by the European Research Council (grant ref: 758813 MHINT). This work was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at the University Hospitals Bristol National Health Service Foundation Trust. PA is an NIHR senior investigator and is funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) Obesity, Diet and Lifestyle Theme and NIHR Oxford and Thames Valley Applied Research Collaboration. JHC has received grants from the National Institute for Health Research Biomedical Research Centre, Oxford, John Fell Oxford University Press Research Fund, Cancer Research UK (grant no: C5255/A18085) through the Cancer Research UK Oxford Centre, and the Oxford Wellcome Institutional Strategic Support Fund (204826/Z/16/Z). JCH is supported by a British Heart Foundation Fellowship (FS/14/55/30806), and acknowledges support from the Oxford Biomedical Research Centre, BHF Oxford Centre for Research Excellence, and Nuffield Department of Population Health. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health and Social Care. We are grateful to the participants of the UK Biobank as well as all the research staff who worked on the data collection and synthesis. This research has been conducted under UK Biobank application numbers 14568 and 40628.
Competing interests PST reports personal fees from AstraZeneca, and personal fees from Duke-NUS, outside the submitted work. CACC reports receiving personal fees from ClinRisk, outside this work. MRM reports grants from Pfizer and Rusan, outside the submitted work. JHC is an unpaid director of QResearch, a not-for-profit organisation which is a partnership between the University of Oxford and EMIS Health. JHC is a founder and shareholder of ClinRisk Ltd and was its medical director until 31 May 2019; ClinRisk produces open and closed source software to implement clinical risk algorithms (outside this work) into clinical computer systems. AvE and JCH work at the Clinical Trial Service Unit and Epidemiological Studies Unit, which receives research grants from industry that are governed by University of Oxford contracts that protect its independence, and has a staff policy of not taking personal payments from industry; further details can be found at https://www.ndph.ox.ac.uk/files/about/ndph-independence-of-research-policy-jun-20.pdf. AKC, HMS, NL and PA have no conflicts to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.