Article Text

Abstract
Objectives Idiopathic pulmonary fibrosis (IPF) primarily affects the aged population and is characterised by failure of alveolar regeneration, leading to loss of alveolar type 1 (AT1) cells. Aged mouse models of lung repair have demonstrated that regeneration fails with increased age. Mouse and rat lung repair models have shown retinoic acid (RA) treatment can restore alveolar regeneration. Herein, we seek to determine the signalling mechanisms that become activated on RA treatment prior to injury, which support alveolar differentiation.
Design Partial pneumonectomy lung injury model and next-generation sequencing of sorted cell populations were used to uncover molecular targets regulating alveolar repair. In vitro organoids generated from epithelial cells of mouse or patient with IPF co-cultured with young, aged or RA-pretreated murine fibroblasts were used to test potential targets.
Main outcome measurements Known alveolar epithelial cell differentiation markers, including HOPX and AGER for AT1 cells, were used to assess outcome of treatments.
Results Gene expression analysis of sorted fibroblasts and epithelial cells isolated from lungs of young, aged and RA-pretreated aged mice predicted increased platelet-derived growth factor subunit A (PDGFA) signalling that coincided with regeneration and alveolar epithelial differentiation. Addition of PDGFA induced AT1 and AT2 differentiation in both mouse and human IPF lung organoids generated with aged fibroblasts, and PDGFA monoclonal antibody blocked AT1 cell differentiation in organoids generated with young murine fibroblasts.
Conclusions Our data support the concept that RA indirectly induces reciprocal PDGFA signalling, which activates regenerative fibroblasts that support alveolar epithelial cell differentiation and repair, providing a potential therapeutic strategy to influence the pathogenesis of IPF.
- idiopathic pulmonary fibrosis
- interstitial fibrosis
Data availability statement
Data are available in a public, open access repository. GEO: GSE157440.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Which epithelial–mesenchymal crosstalk pathways are activated by retinoic acid (RA) pretreatment of aged lungs that support realveolarisation after partial pneumonectomy surgery?
What is the bottom line?
Increased platelet-derived growth factor subunit A (PDGFA)/platelet-derived growth factor receptor A signalling in aged lungs promotes regenerative activation of interstitial matrix fibroblast, which is required for alveolar type 2 to alveolar type 1 (AT1) differentiation and alveolar regeneration.
Why read on?
In vitro and in vivo analyses demonstrated that PDGFA signalling supports alveolar matrix fibroblast and AT1 epithelial cell differentiation, both necessary for alveolar regeneration in aged lungs.
Introduction
Several lung diseases are associated with advanced age, such as lung cancers, COPD and end-stage adult interstitial lung diseases (ILDs), including idiopathic pulmonary fibrosis (IPF). IPF is characterised by extensive fibrosis, hyperproliferation of abnormal alveolar type 2 cells (AT2) and loss of alveolar type 1 cells (AT1), causing progressive respiratory decline and mortality usually within 5 years of diagnosis.1–3 While the pathogenesis of IPF remains unclear, chronic alveolar epithelial cell injury and fibrotic activation of fibroblasts are linked to the disorder.4 However, an understanding of the epithelial–mesenchymal crosstalk during chronic epithelial injury and in the context of ageing remains limited.
Reciprocal communication between epithelial and mesenchymal cells drives lung branching morphogenesis and epithelial differentiation during lung development and repair.5–7 Single-cell RNA sequencing (RNA-Seq) analyses have provided more clarity regarding the heterogeneity of normal epithelial and mesenchymal cells during development, including identifying several distinct heterogeneous pulmonary fibroblast populations.7 8 Of all the pulmonary stromal cells, mesenchymal alveolar niche cells (MANCs)7 and LGR5+ resident alveolar fibroblasts9 have received recent attention due to their capacity to support the alveolar niche and differentiation of the alveolar epithelium. When used in alveolar organoid cultures, MANCs support large organoid formation with high AT1/AT2 ratios. MANCs are Wnt-responsive AXIN2+ cells that also express platelet-derived growth factor receptor A (PDGFRA) and transcriptionally overlap with LGR5+ resident alveolar fibroblasts. In addition to the WNT2+/PDGFRA+ MANCs, there are AXIN2+/PDGFRA+, PDGFRA+ and AXIN2+ resident fibroblast populations.7 An important role of these PDGFRA+ fibroblast populations in normal septation, realveolarisation and bleomycin-induced fibrosis has been demonstrated by multiple previous studies.10–19 Interstitial PDGFRA+ fibroblasts are a mixed population of three functionally distinct fibroblast stages: contractile myofibroblasts, matrix synthesising and remodelling matrix fibroblasts10 11 14 15 and lipid-storing lipofibroblasts.20 21 How these functional stages correlate with platelet-derived growth factor receptor A (PDGFRA), AXIN2 and WNT2 expression remains unclear as marker genes for functional myofibroblasts, matrix fibroblasts and lipofibroblasts such as Acta2, Perilipin and Elastin have overlapping expression in both MANCs and other PDGFRA+ fibroblasts.7 Myofibroblasts, which express low levels of Pdgfra, induce smooth muscle actin expression during postnatal secondary septation and during alveolar regeneration in young adult mice.14–17 Induction of the contractile function is important for the initiation and formation of secondary septa; however, overabundant myofibroblast induction in aged mice results in the failure to regenerate alveolar septa after partial pneumonectomy (PNX).22–24 Moreover, these activated myofibroblasts contribute to bleomycin-induced fibrosis.19 Developmental and regeneration studies suggest that interstitial myofibroblast transitions to a matrix synthesising and remodelling fibroblast stage to stabilise the newly formed septa.14 15 25 In some lung realveolarisation models, retinoic acid (RA) reinitiates myofibroblast activation and septation in young adult rats and mice.16 26 27 In this study, we used preconditioning of aged mice with RA treatment before PNX to understand the downstream mechanisms that result in regenerative activation of fibroblasts, leading to alveolar repair with advanced age. We demonstrate that RA indirectly reinitiates alveolar septation by reducing myofibroblast activation and increasing the activation of matrix fibroblast function. Our in vitro experiments support the idea that PDGFRA-expressing matrix and myofibroblasts are fibroblast activation stages rather than distinct lineages as PDGFA supplementation activates the matrix function. We used gene expression studies after PNX and in vitro organoid models to further study the role of matrix fibroblast in alveolar reseptation and alveolar epithelial differentiation, both important functions for a ‘regenerative activation’ of interstitial lung fibroblasts.
One important problem with tissue fibrosis as a therapeutic target is that it is a late event in the natural history of IPF. By the time fibrotic tissue deposition is sufficient to cause symptoms prompting the patient to seek medical attention, disease progression has advanced to a stage where parenchymal lung fibrosis cannot be repaired in human lungs. As explanted lungs are end-stage disease, we used the organoid model to identify molecular events that occur earlier in the pathogenesis of IPF and are potentially reversible, in contrast to temporally late events such as fibrotic tissue derangement, which is irreversible. In this study, we have focused on the matrix fibroblast stage that is lost with advanced age and supports proper AT2/AT1 differentiation. We evaluated primary alveolar epithelial cells and fibroblasts isolated directly from patients with IPF; we studied live cells in an environment permitting them to communicate with and influence each other, and we identified PDFGA signalling as an important reciprocal messenger to allow for proper AT2/AT1 cell differentiation. These studies provide insights into future treatment options for lung regeneration and early potentially reversible interventions in IPF.
Materials and methods
Mouse husbandry and left lobe PNX
Wild-type mixed background mice were used for flow cytometry analyses and organoid culture. Young mice were 12–16 weeks of age, and aged mice were >40 weeks of age. Based on our previous studies on RA and alveolar regeneration,16 we treated aged mice (Aged RA) with trans-RA (Sigma R2625) at a dose of 2 µg/g body weight dissolved in dimethyl sulfoxide and peanut oil administered via intraperitoneal injection daily for 10 days prior to pneumonectomy with a 2-day break after the first 5 days. Mice were then subjected to PNX or sham surgery (Sham) and harvested 5 or 21 days after surgery.28 For each experimental group (Young Sham, Young PNX, Aged PNX and Aged RA pretreated), n>3 were collected for each experiment.
Tissue harvest
Prior to sacrifice, mice were lethally injected with ketamine, xylazine and acepromazine. For optimal lung histology, lungs were inflation-fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) at 25 cm H2O pressure via a tracheal cannula and closed chest to ensure comparable histology of fixed tissue. Lungs were fixed overnight at 4°C. All lungs were washed with PBS, dehydrated through a graded series of ethanol solutions and processed for paraffin embedding. Sections (5 µm) were loaded onto polylysine-coated slides for analysis. All samples were processed at the same time to ensure no difference between Sham and PNX-operated mice. Use of animals was approved by Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Animal Care and Use Committee.
Morphometric point and intersection counting analysis
As in previously published studies, we used morphometric point intersection analysis to quantify alveolar simplification of fractional airspace area on histological sections of inflation-fixed adult lungs at 21 days after PNX.15 16 For each animal, three to five sections from different parts of the lung, showing all five lobes, were analysed. A 120-point grid was overlaid over five to seven random pictures of each lobe. The alveolar space was calculated as the percentage of grid intersections over the alveolar space versus the alveolar tissue. Each symbol represents the average percentage of airspace of one animal. Values for each individual animal were averaged per experimental group.29
Patient samples
Donor patient samples were obtained from healthy lungs rejected for transplant, and the CCHMC Institutional Review Board declared that donor tissue samples were Institutional Review exempt, in accordance with protocol 2013-3356. Samples were collected from explanted lungs of patients with ILD at the time of lung transplant, and informed consent was obtained from each subject in accordance with the Partners Institutional Review Board (2013P002332), Boston, Massachusetts, USA. Patient clinical data are available in online supplemental table S6.
Supplemental material
Second harmonic generation and immunofluorescence
IPF and donor-fixed lung tissue in optical cutting temperature (OCT) blocks were sectioned at 250 µm and cleared via the passive clarity (PACT) protocol, followed by whole-mount antibody stain.30 In short, OCT is removed with PBS, and then the tissue is placed in a cold 4% polyacrylamide (1610140, Bio-Rad) hydrogel with 0.25% photoinitiator (VA-044, Wako) solution overnight. The tissue was polymerised at 37°C for 4 hours, washed with PBS to remove excess hydrogel and then incubated at 37°C overnight in an 8% sodium dodecyl sulfate in PBS solution for permeabilisation. After washing with PBS, a 25% Quadrol (122262-1L, Sigma Aldrich) solution in PBS was added for 16 hours on a 37°C rotator, after which the samples were washed in PBS and are ready for immunostaining. For immunostaining, the PACT-cleared tissue sample was incubated with 4% donkey serum blocking solution at room temperature and then incubated with primary antibodies aSMA (Sigma A5228) and Abca3 (Seven Hills WMAB-17G524) at 4°C for 3 days. Unbound antibody was removed with PBS, followed by fluorescent labelling with secondary antibodies. After washing with PBS, the tissue was mounted in refractive index matching solution (RIMS) solution for second harmonic generation (SHG) imaging on a Nikon FN I upright microscope and analysed using Nikon Elements and Imaris software.
Cell sorting and fluorescence activated cell sorting (FACS)
Cell sorting was performed using Miltenyi Biotec’s MicroBead positive selection. Human or mouse epithelial cellular adhesion molecule (EPCAM or CD326+) antibody-conjugated beads were used to isolate epithelial cells, and human or mouse PDGFRA-conjugated (CD140+) beads were used to isolate PDGFRA+ fibroblasts. Isolation was performed as per manufacturer’s instructions using LS columns for positive selection. Flow cytometry was performed using fluorescent-labelled antibodies (online supplemental table S7). Cells were sorted using a Becton Dickinson LSRII analyser with five lasers. Proliferation was determined by Ki67+ expression in CD326+, CD140+ and CD140+CD34+ cell populations.
Supplemental material
RNA analysis (quantitative PCR and RNA-Seq)
RNA-Seq was performed by CCHMC’s Gene Expression Core. The resulting fastq files were aligned to mm10 or GRCh37 using Bowtie2.1 Raw gene counts were obtained using Bioconductor’s Genomic Alignment, and normalised FPKM (fragments per kilo base per million mapped reads) values were generated using Cufflinks.2 4 Mouse RNA-Seq was performed on epithelial cellular adhesion molecule (Epcam) EPCAM+ or PDGFRA+ lung cells from mice aged 12–16 weeks following PNX surgery and in >12-month-old mice following Sham, PNX surgery or PNX surgery with RA pretreatment (n=3). Whole mouse lungs were dissociated using dispase (Corning) and sorted with CD140 or CD326 microbeads (Miltenyi Biotec) before sequencing. Human donor and IPF samples were also dissociated and microbead-sorted similar to mouse samples for CD140+ before sequencing (n=3). DESeq was used to analyse raw gene counts and calculate differentially expressed genes.3 To identify differentially expressed genes in mice, a cut-off of fold -change (FC) FC >2 and p value <0.01 was used, and a cut-off of FC >1.5 and p value <0.05 was used for human samples. All differentially expressed genes were expressed with an FPKM >1 in at least half of the replicates in one of the conditions being compared. Mouse RNA-Seq gene expression patterns were determined by analysing all genes differentially expressed after aged PNX or after aged RA pretreatment PNX when compared with aged Sham. These differentially expressed genes were used to make a z-score-normalised FPKM expression matrix using all samples. Partek Genomics Suite’s hierarchical clustering coupled with heatmap generation was then used to cluster and visualise expression patterns. ToppGene’s ToppFun was used to identify functional enrichment hits of significantly altered RNAs within particular patterns.5 Upstream driver analyses were performed using differentially expressed genes from a given comparison using Ingenuity Pathway Analysis’ upstream analyses software. Reverse transcription PCR (RT-PCR) was performed by generating complementary DNA (cDNA) using iScript (Bio-Rad). TaqMan assay probes (Thermo Fisher) were used to assess RNA expression, and probes used are tabulated in online supplemental table S8.
Supplemental material
Organoid generation
Human and mouse lung tissue were dissociated into single-cell suspensions using dispase and DNase. Cell suspensions were incubated with fragment crystallizable receptor (FcR) blocking reagent in magnetic-advanced cell sorting (MACS) MACS buffer (Miltenyi Biotec), followed by isolation of PDGFRA (CD140+) and EPCAM (CD326+) cells as described earlier. PDGFRA (CD140+) fibroblasts were co-cultured with EPCAM (CD326+) cells in a ratio of 10:1, which was determined and optimised in pilot studies based on previously published data.31 32 Mixed cells were combined with Matrigel (laminin, coll-iv, entactin) in a 1:1 ratio and cultured on a transwell insert in 24-well plates and incubated at liquid–air interface in MTEC plus media (Dulbeccos Modified Eagle Medium-Ham’s F-12, HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), penicillin and streptomycin, fungizone, insulin, transferrin, cholera toxin, epithelial growth factor (EGF) and bovine pituitary extract). Organoid cultures were grown for 21 days and then fixed for immunofluorescence analysis. Murine organoids were generated by a pool of three to six mice for independent experiments, resulting in three to six technical replicates, and repeated more than three times. Human organoids were generated from two donors (>50 years): five patients with IPF and one patient with ILD (51–70 years). It is important to note that for RA-treated cells, RA was not used in the organoid cultures but was administered to the mice 2 weeks before PNX surgery and cell harvest to generate organoids.
Statistics
Statistical analysis of two-variable quantitative PCR (qPCR) experiments was assessed by non-parametric Mann-Whitney test. For all qPCR and immunofluorescence quantification with more than two variables, statistics were determined by non-parametric analysis of variance (ANOVA), followed by Kruskal-Wallis multiple comparisons test comparing each column. P values shown on graphs compare each column with the respective control column. Values of p<0.0001 are denoted with ***.
Results
To identify the role of resident PDGFRA+ interstitial fibroblasts in repair of the aged lung, we used the murine PNX model and compared alveolar regeneration in mice aged 12–16 weeks (hereafter referred to as ‘young mice’) with mice aged >40 weeks (hereafter referred to as ‘aged mice’).23 Consistent with previously published data, regeneration in aged mice is decreased and incomplete by 21 days after surgery23 24 (figure 1A). As RA reinitiates septation in other lung regeneration models,16 26 27 we hypothesised that the treatment of aged mice with RA prior to PNX injury (hereafter referred to as ‘RA pretreatment’) would promote alveolar regeneration in aged lungs. Male and female aged mice were treated with RA for 10 days prior to PNX surgery. Lungs from these mice were analysed 5 and 21 days after surgery using histology and flow cytometry. RA pretreatment restored alveolar septation in aged mice (figure 1A,B). We have previously shown that myofibroblast and matrix fibroblast orchestrate alveolar regeneration in young mice and that myofibroblast co-expressed PDGFRA (CD140) and CD29, whereas matrix fibroblast co-expressed PDGFRA (CD140) and CD34.15 Young mice actively regenerate their lung after PNX, with increased proliferation of CD326+ epithelial cells compared with young sham mice. While epithelial proliferation (CD326) is not changed in aged mice after PNX, proliferation of CD140/CD34 matrix fibroblast is reduced in aged mice compared with young mice (p=0.011). However, there was no significant difference in proliferation in CD140/CD34 matrix fibroblasts between regenerating young and regenerating RA pretreatment of aged mice (p>0.99), suggesting a restored proliferation of CD34-positive fibroblasts (figure 1C–E). Overall, the number of PDGFRA-expressing cells was similar between young and aged lungs. These data suggest that matrix fibroblast proliferation plays an important role in alveolar repair.
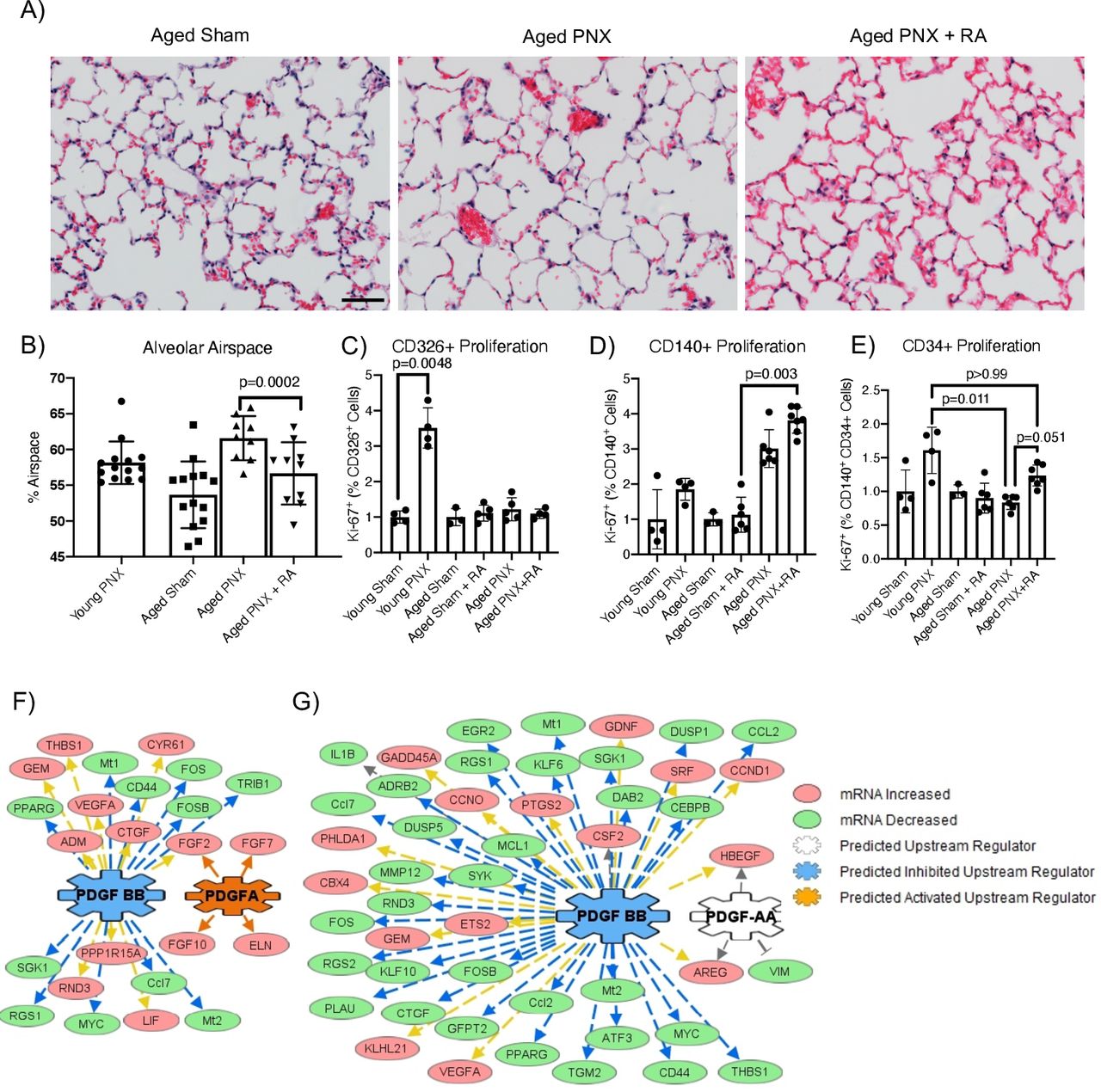
Mouse lung regeneration following RA pretreatment. (A) H&E stain of aged Sham, aged mice and RA-pretreated mice 21 days after PNX. Scale bar is 200 µm. (B) Quantification of alveolar space by morphometric point and intersection counting analysis (PNX21). (C–E) Proliferation was assessed by KI67 expression by flow cytometry (PNX5). Data were normalised to respective sham controls. (C) CD326+, (D) CD140+ and (E) CD140+CD34+ cells in young and aged Sham, PNX and RA pretreatment mice. (F, G) Upstream analyses of RNA sequencing gene expression changes in PDGFRA+ mesenchymal cells from ‘aged mice’ when compared with either ‘young mice’ or ‘RA pretreatment’ mice (PNX5). In both comparisons, genes downstream of PDGF signalling were significantly altered. (F) In PDGFRA+ mesenchymal cells isolated from RA pretreatment, PDGF-BB signalling was predicted to be significantly inhibited, while PDGFA signalling was activated. (G) Similarly, in young mice compared with aged mice, PDGF-BB signalling was predicted to be significantly inhibited, with PDGFA signalling being altered in PDGFRA+ mesenchymal cells. mRNA, messenger RNA; PDGF, platelet-derived growth factor; PDGFA, platelet-derived growth factor subunit A; PNX, partial pneumonectomy; RA, retinoic acid.
RNA-Seq of PDGFRA (CD140+) fibroblasts and EPCAM+ (CD326) epithelial cells isolated from sham, young, aged and RA pretreatment mice was used to identify changes in gene expression associated with alveolar regeneration 5 days after PNX surgery (online supplemental tables 1 and 2). Based on RNA-seq expression data, both PDGFRA and PDGFA were significantly upregulated in fibroblasts and epithelial cells in the lungs of RA-pretreated mice compared with aged lungs. Changes in gene expression patterns between PDGFRA+ fibroblasts and epithelial cells were strikingly different. In aged PDGFRA+ fibroblasts, a large subset of genes are altered by PNX, while RA pretreatment prevented these changes (online supplemental figure 1A). In contrast, in the epithelial cells, RA pretreatment induced a large subset of genes that were not changed by PNX (online supplemental figure 1B). These data suggest that PNX in aged lungs induces ‘misdirected’ gene activation and inactivation of PDGFRA+ fibroblasts that result in failure to regenerate. RA prevents these ‘misdirected’ gene changes, allowing regeneration. In the epithelium of aged lungs, PNX does not induce gene changes, but RA pretreatment promotes gene changes that result in regeneration.
Supplemental material
Supplemental material
Functional enrichment analysis of the ‘misdirected’ gene changes in PDGFRA+ fibroblasts shows that RA pretreatment prevented the induction of genes associated with apoptosis, chromatin remodelling and inflammation while simultaneously preventing the loss of genes associated with matrix organisation, mesenchymal cell differentiation, lung development, stem cell differentiation and regulation of epithelial cell proliferation (online supplemental figure 1C). In contrast, functional enrichment analysis of the gene changes in epithelial cells shows that RA pretreatment inhibited the expression of genes associated with inflammation and cytokine production, while genes associated with epithelial development, branching, proliferation and differentiation were induced (online supplemental figure 1D). These data demonstrate that in both PDGFRA+ fibroblasts and epithelial cells, inflammation suppression coupled with induction of cell-type-specific differentiation is required to allow for alveolar regeneration.
To identify gene networks associated with alveolar regeneration in PDGFRA+ fibroblasts, upstream driver analysis was performed using the gene changes observed between RA pretreatment and aged PDGFRA+ fibroblasts (online supplemental table 3). Active PDGFA signalling and inactive PDGF-BB signalling were predicted as important regulators of gene changes that allow realveolarisation (figure 1F). Similarly, in young mice compared with aged mice (online supplemental table 4), inactivation of PDGF-BB signalling and alteration of PDGF-AA were predicted to be primary mediators of the alveolar regeneration process (figure 1G). Both PDGFA and PDGFB are expressed in the respiratory epithelium, suggesting paracrine signalling from the epithelium to the resident interstitial fibroblasts. RNA-Seq analysis of EPCAM-sorted cells identified a significant 5.9-fold increase in PDGFA expression in aged PNX+RA compared with aged PNX. RT-PCR was used to assess levels of Pdgfa in epithelial cells isolated from sham, young, aged and aged RA pretreatment lungs but showed no statistical difference due to small sample size (online supplemental tables 1 and 2; online supplemental figure 1E). Taken together, these data suggest that increased reciprocal PDGFA signalling is essential for alveolar regeneration after PNX.
To test the role of PDGFA signalling in supporting alveolar regeneration and epithelial differentiation, an epithelial–mesenchymal mixed cell organoid system was used. Lung organoids were generated by combining primary EPCAM+ lung epithelial cells with primary PDGFRA+ fibroblasts isolated from young, aged and RA-pretreated murine lungs. Alveolar epithelial cell differentiation was assessed by immunofluorescence staining with SFTPC (AT2 cells), HOPX and AGER (AT1 cells) and quantified by morphometry (figure 2A,B). SFTPC, AGER and HOPX expression in primary lung epithelial cells from aged mice was increased when co-cultured in organoids with young or RA-pretreated mouse lung PDGFRA+ fibroblasts, compared with organoids co-cultured with PDGFRA+ fibroblasts from aged mice. Independent of age and RA pretreatment, epithelial cells showed no difference in AT2/AT1 differentiation when cultured with young PDGFRA+ fibroblasts. While RNA-Seq demonstrated that epithelial cells from young and RA-pretreated mice expressed higher levels of Pdgfa, their ability to differentiate into AT2 or AT1 cells was impaired when cultured with aged PDGFRA+ fibroblasts. When organoid cultures from aged PDGFRA+ fibroblasts were supplemented with PDGFA ligand, epithelial AT2/AT1 differentiation was restored as the expression of AT1 cell markers, HOPX and AGER, was increased (figure 2B). Taken together, these data demonstrate that the ‘aged’ PDGFRA+ cells have lost the ability to promote epithelial differentiation in organoid culture and that ‘aged’ epithelial cells still retain the potential to differentiate into AT2/AT1 cells.

RA pretreatment and PDGFA treatment support alveolar differentiation in mouse lung organoids. (A) Immunofluorescence analysis of SFTPC, HOPX and AGER in mouse lung organoids generated with young epithelium and aged PNX fibroblast or aged epithelial and aged fibroblast treated with PDGFA after 21 days in culture. (B) Quantification of total area masked by staining compared with total area of DAPI (AGER) or total cell counts as a percentage of total DAPI (4′,6-diamidino-2-phenylindole) nuclei (HOPX and SFTPC) for the different combinations of mouse lung organoids generated demonstrates increased AGER and HOPX expression in organoids generated with young or aged RA-pretreated fibroblast and PDGFA-treated organoids generated with aged fibroblasts. SFTPC expression was increased in organoids generated with RA-pretreated aged fibroblasts. *** indicates p<0.0001 compared with organoids generated with aged fibroblasts. (C) Quantification of RNA expression changes from IMR90 cells treated with PDGFA ligand demonstrated increased matrix fibroblast differentiation. PDGFA, platelet-derived growth factor subunit A; PNX, partial pneumonectomy; RA, retinoic acid.
IMR90 human fetal lung fibroblasts express PDGFRA. To determine the role of epithelial PDGFA-positive paracrine feedback loop on Pdgfra expression and matrix fibroblast differentiation, IMR90 cells were treated with PDGFA ligand. PDGFA treatment increased the expression CD248, WNT5A, FGF1 and PDGFRA, previously identified matrix fibroblast signature genes, and reduced the expression of WNT2 and ACTA2, previously identified myofibroblast signature genes8 14 (figure 2C). These data support the concept that epithelial PDGFA signalling induces a positive paracrine feedback loop with PDGFRA+ fibroblasts and promotes matrix fibroblast differentiation, which in turn support epithelial AT2/AT1 differentiation.
IPF is associated with advanced age, failure of alveolar repair and impaired AT1 differentiation. Based on the role of PDGFRA+ fibroblast in AT1 differentiation, we therefore examined PDGF signalling in IPF. Single-cell analyses of the IPF and donor epithelium identified subsets of epithelial cells that produce high levels of PDGFB and PDGFA, with PDGFB signalling only being detected in IPF epithelial cells.6 Due to altered PDGF expression in IPF lungs, we hypothesised that the interstitial lung fibroblast population would be altered in IPF lungs. To assess the fibroblast populations’ composition changes in patients with IPF, peripheral tissue of age-matched donor lungs and non-fibrotic peripheral areas of IPF lungs were digested into single-cell suspensions and subjected to flow cytometry. Flow cytometry revealed a loss of 90% of CD140+ matrix fibroblasts in IPF (online supplemental figure 2A). RNA-Seq of CD140+ fibroblasts from non-fibrotic areas of IPF lungs revealed changes in gene expression related to ‘extracellular matrix organisation’, ‘response to wounding’ and altered ‘chromatin assembly’, when compared with donor lung CD140+ fibroblasts (online supplemental figure 2B,C and online supplemental table 5). Furthermore, upstream regulator analysis of differentially expressed genes in IPF-derived CD140+ fibroblasts predicted the activation of PDGF-BB ligand homodimer signalling and reduced PDGF-AA ligand homodimer signalling in IPF, which is consistent with the finding in aged PNX-injured mouse lungs (online supplemental figure 2D). Together, these data suggest a loss of normal interstitial matrix fibroblast as an additional pathological feature of IPF.
Supplemental material
Recent proteomics analysis of young and aged murine lungs33 described downregulation of collagen XIV, which integrates collagen bundles by binding to collagen I fibrils and decorin.34 Changes in extracellular matrix composition and distribution have been previously suggested in aged and IPF lungs.35 To assess whether the loss of matrix fibroblast in non-fibrotic areas of the IPF lungs also results in changes in the extracellular matrix, we performed SHG resonance imaging to visualise collagen structures and immunofluorescent analysis of smooth muscle actin and AT2 cell distribution in alveolar regions of normal (n=3) and IPF (n=3) lungs (figure 3A). Normal lungs showed an intricate collagen network spanning the lung parenchyma and supporting alveolar structures.

IPF lungs contain abnormal fibroblast populations. (A) SHG resonance imaging (green) showing the structure of collagen in donor, ‘less severe’ region of IPF and ‘severe’ region of IPF lungs co-stained with immunofluorescence labelling of ABCA3+ AT2 cells (red) and aSMA+ smooth muscle (white), in 250 µm thick lung sections. Scale bars indicate 50 µm. RA pretreatment and PDGFA treatment enhance the expression of alveolar epithelial cell differentiation in human lung organoids. (B–C) Immunofluorescence analysis of AGER, HOPX and SFTPC demonstrates increased expression of IPF epithelial cells cultured with aged mouse fibroblasts or treated with PDGFA. (D) Quantification of area imaged with AGER expression as normalised to total DAPI area. HOPX-positive and SFTPC-positive cells are normalised to total DAPI nuclei. Oganoids generated with IPF epithelium and young mouse fibroblasts expressed less SFTPC (p=0.0003), AGER (p=0.007) and HOPX (p=0.031) compared with organoids generated with normal donor epithelial cells. *** denotes p<0.0001 compared with organoids generated with aged fibroblasts. IPF, idiopathic pulmonary fibrosis; PDGFA, platelet-derived growth factor subunit A; RA, retinoic acid; SHG, second harmonic generation.
We determined non-fibrotic areas with apparently normal alveolar structures in IPF lungs by H&E and performed SHG imaging on adjacent sections (online supplemental figure 2). SHG revealed that the intricate collagen structure found in donor lungs was amorphous, suggesting impaired support of alveolar structures. Dense collagen structures and considerable smooth muscle actin replace alveolar structures in fibrotic foci (figure 3A). These data suggest that the loss of matrix fibroblast function in the IPF lung contributes to destruction of the collagen network, revealing a possible underlying early pathological change within the extracellular matrix in IPF lungs.
IPF is characterised by the loss of AT1 cells, but it remains unclear whether this is due to a defect of AT2 cells to differentiate and replenish chronically stressed AT1 cells.1 Our data showed the loss of matrix fibroblasts in IPF and suggest that matrix fibroblasts are required for AT1 differentiation (figure 3 and online supplemental figure 2). To test the hypothesis that loss of AT1 cells in IPF is due, at least in part, to loss of matrix fibroblasts, we assessed AT1 cell differentiation in human/mouse organoids. Organoids were generated by combining human CD326+ epithelial cells isolated from normal donor and IPF lungs with murine young, aged or RA-pretreated PDGFRA+ fibroblasts. We assessed AT2/AT1 differentiation in human donor or IPF epithelial cells with immunofluorescence analysis of HOPX, AGER and SFTPC. While characteristic morphological features of AT2 and AT1 cells were not readily visible, protein expression of cell-type-specific markers was discernible. Only young and RA-pretreated fibroblasts supported AT2/AT1 differentiation in organoids generated from both donor and IPF epithelial cells (figure 3B,D). Organoids generated with IPF CD326+ cells expressed reduced AGER, HOPX and SFTPC compared with organoids generated with donor CD326+ cells co-cultured with young CD140+ cells. Other epithelial markers (P63, MUC5B, acetylated tubulin and CCSP) were comparable among all combinations of fibroblast and epithelial cells, indicating there was no shift in the bronchoalveolar/bronchial organoid ratio (online supplemental figure 4).
Supplemental material
PDGFA treatment of organoids generated from aged murine fibroblasts and human IPF epithelial cells enhanced AT1 (HOPX and AGER) and AT2 (SFTPC) cell differentiation to similar levels as organoids generated with RA-pretreated or young fibroblasts (figure 3C,D). These data suggest that epithelial cells from patients with IPF have not entirely lost the ability to differentiate into AT1 cells and that fibroblasts with active PDGFA/PDGFRA signalling are required for AT1 cell differentiation in IPF.
To further test the importance of PDGFA signalling, aged mouse epithelial cells were co-cultured with young, aged or RA-pretreated PDGFRA+ fibroblasts and either treated with PDGFA-neutralising antibody to directly inhibit PDGFA activity or treated with nintedanib, a pan tyrosine kinase inhibitor, to block all PDGF (PDGFRA, PDGFRB and PDGFRC) receptors, among other pathways. PDGFA-neutralising antibody treatment reduced AGER, HOPX and SFTPC expression in organoids generated with young and aged RA-pretreated PDGFRA+ fibroblasts (figure 4A). Organoids generated with aged PDGFRA+ fibroblasts had low expression of AGER and HOPX; however, treatment with nintedanib enhanced the expression of these AT1 markers (figure 4B). These results demonstrate that specific inhibition of PDGFA ligand is sufficient to reduce AT1 differentiation. In contrast and consistent with previous publications, the pan tyrosine kinase inhibitor nintedanib increases AT1 cell differentiation in aged organoids, suggesting a ratio of PDGFA/PDGFB homeostasis is needed to support epithelial cell differentiation. However, in organoids, direct activation of PDGFA is sufficient to support alveolar differentiation, whereas inhibition of PDGFA is sufficient to block AT1 cell differentiation.

Inhibition of PDGFA blocks alveolar differentiation. Organoids generated with aged mouse epithelium co-cultured with young, aged and RA-pretreated fibroblasts. Organoids were treated with control, PDGFA-neutralising antibody or nintedanib. (A) Immunofluorescence analysis demonstrates decreased expression of AGER and HOPX in organoids generated with young or RA-pretreated fibroblasts following PDGFA antibody treatment. Nintedanib increased the expression of AGER and HOPX in organoids generated with aged fibroblasts. (B) Quantification of AGER (normalised to total DAPI area) and HOPX (normalised to total DAPI-positive cell number) expression. PGDGFA, platelet-derived growth factor subunit A; RA, retinoic acid.
Discussion
Potential therapeutic advantage of PDGFA ligand treatment
The current focus of IPF research has revolved around correcting alveolar regeneration caused by chronic epithelial cell injury which is believed to be an underlying cause of the disease. The US Food and Drug Administration (FDA) has approved two drugs, pirfenidone and nintedanib, a broad tyrosine kinase inhibitor that acts on several targets including PDGFRA and PDGFRB. Thus far, these FDA-approved treatments slow loss of forced vital capacity but have not significantly enhanced life expectancy following diagnosis.36 37 Several recent clinical trials (phases 2 and 3) have continued along this path, working to block other aberrant epithelial signalling pathways (mTOR, YAP, TGFβ) identified in IPF.5 38 However, perhaps a return to the molecular and cellular aspect of the diseased fibroblast would promote the identification of new targetable treatment options.39 Fibroblasts support alveolar regeneration, and previous findings demonstrated RA pretreatment enhanced alveolar regeneration in animal models,26 27 but RA treatment did not enhance alveolar repair in clinical trials treating COPD.40 Similarly, direct treatment of mouse organoids with RA, by Ng-Blichfeldt et al, resulted in smaller organoid size with reduced epithelial cell differentiation, whereas inhibition of RA increased organoid size and increased epithelial cell proliferation via activation of YAP and FGF signalling.41 In our study, organoid cultures were not treated with RA, but mice were administered RA 2 weeks prior to PNX surgery and cell harvest. Together, these data suggest RA has a positive preconditioning effect but is inhibitory during active regeneration. RA treatment of patients with IPF would be after disease diagnosis and onset; thus, it is very unlikely to be effective. RA is an epigenetic modulating agent that is extensively used in reprogramming fibroblast to induced pluripotent stem cells. With advanced age, epigenetic modifications increase. Understanding the cellular and molecular mechanisms of induction of RA-mediated regeneration in aged lungs could be central for effective translation into elderly patients with lung disease and may reveal novel insights into the pathogenesis of alveolar disease and senescence.42 In this study, we used RA pretreatment prior to PNX injury to understand the downstream mechanisms that result in regenerative activation of fibroblasts, leading to alveolar repair with advanced age.
A large number of gain-of-function and loss-of-function mutations in PDGF and PDGFR genes have been generated and demonstrated that the PDGF/PDGFR plays important roles in the development and that PDGF overexpression or misexpression may induce pathological responses.43 While loss of PDGFA signalling during lung development resulted in failure to form secondary septa, misexpression of PDGFA in the pulmonary epithelium resulted in overproliferation of mesenchymal cells compromising the expansion of distal airspaces.12 44 45 PDGFA and PDGFC are potent inducers of mesenchymal proliferation, whereas PDGFB is more pleiotropic and induces an inflammatory response.43 PDGFA binds PDGFRA, which is mainly expressed in mesenchymal cells in the lung and has been detected in subtypes of mesenchymal progenitors in lung, skin and intestine, in a homodimer or PDGFA/PDGFB heterodimer. PDGFRB is expressed in mesenchyme, particularly in vascular smooth muscle cells and pericytes.46 Pulmonary hypertension, lung cancer and lung fibrotic diseases have been linked with elevated PDGF signalling. Murine models of lung fibrosis indicated elevated PDGFB signalling as the major source of inflammation and fibrotic response with the role of PDGFA signalling not been extensively studied.47
Our current findings support the notion that the activation of PDGFA signalling mediated by RA treatment before injury resulted in gene expression changes in fibroblasts that promote epithelial regeneration. In vitro treatment of primary rat lung fibroblasts with RA increased the expression of Pdgfra.48 We found a similar increase in Pdfgra expression in lung fibroblasts of RA-pretreated mice. We found that the epithelium of RA-pretreated mice produces increased Pdgfa ligand after injury. Bronchoalveolar organoids generated with aged mouse CD140+ fibroblasts fail to differentiate both AT2 and AT1 cells in vitro regardless of the types of epithelial cells used. However, treatment of these organoids with PDGFA ligand supports alveolar epithelial cell marker expression, suggesting PDGFA is sufficient to induce alveolar differentiation. PDGFA ligand appears to activate a cascade of epithelial–mesenchymal crosstalk, likely through enhanced fibroblast differentiation into beneficial matrix fibroblasts, which promotes alveolar regeneration. Further studies identifying the underlying cause of loss of the PDGFRA+ matrix fibroblast population in aged and IPF lungs could provide future therapies for patients with IPF in need of efficacious treatment.
The ageing lung
Age is associated with increased risk of several diseases including IPF. Single-cell RNA-Seq analysis of aged murine lungs revealed altered transcriptional signalling that was associated with epigenetic changes in multiple cell types.33 RA is known to place or remove epigenetic marks on histones and DNA.49 In this study, we demonstrate that RA pretreatment prevented many gene expression changes associated with aged fibroblasts and induced epithelial cell gene expression.
It has been previously shown that monocyte recruitment is important for realveolarisation after partial PNX.50 Based on single-cell RNA-Seq analysis, aged fibroblasts have an increased proinflammatory signature.33 Functional enrichment analysis in this study shows that RA pretreatment prevented the induction of genes involved in inflammation in both fibroblasts and epithelial cells. Taken together, these data suggest that aged lungs are predisposed to inflammation and that inflammation induced by PNX needs to be controlled to allow regeneration.
This study demonstrates that pulmonary fibroblast plays an important role in alveolar repair, which is lost with age, and highlights the ability of aged epithelium or epithelium from patients with IPF to differentiate into AT1 cells in the presence of regenerative matrix fibroblast. Our flow cytometry and RNA-Seq data demonstrate that in IPF the PDGFRA+ fibroblast population is reduced and shifted toward myofibroblast differentiation; to our knowledge, similar flow cytometry data have not been published. Single-cell seq data on IPF lungs support the reduced presence and expression of PDGFRA in lung fibroblasts (http://www.IPFcell.atlas).35 51–53 Our in vitro data in IMR90 cells demonstrate that PDGFA supplementation shifted the PDGFRA-expressing cells towards the matrix functional stage. Similar shift to matrix function has been shown by activation of PDGFRA signalling by a constitutive PDGFRA mutation.14 54 With age, PDGFA signalling is reduced in mouse epithelium. Direct supplementation of PDGFA in IPF human/mouse organoids can induce IPF epithelial cell AT1 cell differentiation. Upstream regulator analysis of disease-related gene changes predicted increased PDGF-BB ligand homodimers and loss of PDGFA ligands in IPF fibroblasts driving disease progression, consistent with previous observations.55 In support of its putative role as a driver of IPF pathogenesis, PDGF-BB ligand homodimers increase fibrosis in other injury models, and blocking PDGF-BB ligand homodimers reduces fibrosis in bleomycin-injured mouse lungs.56 These findings, coupled with our PDGFA-neutralising antibody and nintedanib treatment data, suggest that there is a balance between the fibrotic response induced by PDGF-BB ligand homodimers and the alveolar repair supported by PDGFA signalling. FDA-approved nintedanib is a broad target tyrosine kinase inhibitor that blocks both PDGFRA and PDGFRB signalling cascades.36 55 56 Our data suggest that specific activation of PDGFA ligand and specific inhibition of PDGFB ligand may further enhance alveolar repair, supporting a balance between PDGFA and PDGFB signalling cascades. Future therapies may rely on a combination of blocking aberrant pathways (PDGFRB) while supporting the activity of essential pathways for alveolar repair (PDGFA).
Organoid model system
Explanted lungs are end-stage disease that provide a very valuable source of live disease tissue that can be used in the organoid model to identify molecular events that occur during the pathogenesis of IPF and to test potential therapeutic targets (ie, abnormal AT2–fibroblast crosstalk). We have focused on the fibroblast stage that supports proper AT2/AT1 differentiation in this study. We evaluated primary alveolar epithelial cells and fibroblasts isolated directly from patients with IPF; we studied live cells in an environment permitting them to communicate with and influence each other, and we identified age-dependent differences in promoting AT2/AT1 cell differentiation.
The present RNA analysis demonstrates that epithelial and fibroblast interactions are required for alveolar regeneration. Both human normal donor and IPF CD326+ epithelial cells have the potential to differentiate into AT2 and AT1 cells, although IPF epithelial cells have reduced ability, suggesting the underlying mechanism for repair is still present in the IPF epithelium. This work suggests that the fibroblast activates PDGFA ligand in the epithelium to promote alveolar regeneration, a finding consistent with other reports showing that the fibroblast plays a key role in alveolar differentiation. In previous studies, PDGFRA-GFP fluorescent label was used to isolate MANCs, the subset of WNT2+/PDGFRA-GFP+ interstitial fibroblasts that interact with alveolar epithelial cells to support repair and development of the alveolar region.7 We have previously published that the PDGFRA-GFP and PDGFRA expression detected by the CD140 antibody does not perfectly overlap.14 15 In this study, we investigate murine and human PDGFRA-expressing cells by isolating them using the PDGFRA antibody (CD140) and not the PDGFRA-GFP fluorescent label; we also do not separate them by expression of Axin2 and/or Wnt2. We and others have previously demonstrated that these PDGFRA+ fibroblasts are a mixed population of three functionally distinct fibroblast stages, myofibroblasts, matrix fibroblasts and lipofibroblasts, with increased population of one or the other subtypes depending on the context of age, regeneration and homeostasis.10 11 14 15 20 21 Previously published studies22–24 and our current study demonstrate that age and injury promote a myo subtype and that in IPF the matrix subtypes are significantly reduced (figure 5). Treatment of freshly isolated rat fibroblasts with RA resulted in upregulation of PDGFRA expression consistent with our finding that RA pretreatment increased PDGFRA expression in CD140+ lung fibroblasts.48 Moreover, preconditioning lungs with RA prior to injury resulted in preventing ‘misdirected’ gene changes in fibroblasts and promoted gene changes in epithelial cells that result in regeneration. These gene expression changes indirectly activated increased Pdgfa and Pdgfra expression, which started a positive paracrine feedback loop between PDGFRA fibroblasts and AT2 cells. Increased PDGFRA signalling has been demonstrated to increase the matrix fibroblast function in PDGFRA fibroblasts.14 54 Our organoid model suggests that an increase in PDGFRA signalling supports AT2 to AT1 differentiation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of fibroblast functional stages and reciprocal interactions with alveolar type 2 (AT2) cells. Age and injury promote a myofibroblast function in PDGFRA+ fibroblasts.23 In this study, we show that in IPF the matrix fibroblast function is impaired. Myofibroblasts are characterised by the expression of PDGFRA/CD29/aSMA, and matrix fibroblasts are characterised by the expression of PDGFRA/CD34/elastin.14 Increased PDGFRA signalling promotes myo to matrix transition.14 54 RA pretreatment increased PDGFA and PDGFRA expression in lung fibroblasts and AT2 cells, resulting in a positive paracrine feedback loop between fibroblasts and AT2 cells. Organoids made from PDGFRA+ fibroblasts isolated from aged mice are predominantly myofibroblasts and support organoid formation lacking alveolar type 1 (AT1) cell marker expression. Organoids made from PDGFRA+ fibroblasts isolated from young or RA-pretreated mice are predominantly matrix fibroblasts and induce increased PDGFRA expression in co-cultured lung epithelial cells, supporting alveolar organoid formation and AT1 cell differentiation. IPF, idiopathic pulmonary fibrosis; PDGFA, platelet-derived growth factor subunit A; PDGFRA, platelet-derived growth factor receptor A; RA, retinoic acid.
These findings demonstrate the need to study alveolar regeneration and differentiation in a non-cell autonomous context. Herein, a relatively simple model of lung development and regeneration was used, in which interstitial fibroblasts and alveolar epithelial cells were co-cultured. This model demonstrates a need not only to address the aberrant alveolar epithelial cells in ILDs such as IPF but also to take into consideration the fibroblasts, and potentially other cell types, that support alveolar remodelling during injury.
Limitations
Our studies were limited by sample numbers. Pneumonectomy is a survival surgery; thus, the number of mice used in each study was adjusted to reduce stress on the animals. Access to patient samples and patient diagnosis was limited to IPF for RNA-Seq and to usual interstitial pneumonia/non-specific interstitial pneumonia and IPF for organoid generation. We used CD326+ selection for our epithelial isolation as others and our previous work demonstrate distal lung epithelial cells do not express ‘normal’ AT2 cell markers. We performed CD326+ isolation on murine lungs as well to be consistent between human and mouse.1 2 57 58 RNA-Seq analysis was unable to assess the signalling mechanism from the mesenchyme to the epithelium that supported AT1 cell expression.
Other study designs and experimental set-ups could overcome this limitation and identify that potential crosstalk mechanisms or alteration of matrix stiffness is possible to support AT1 cell differentiation.7 59 60 Our analysis demonstrates that PDGFA is increased in the epithelium, and addition of PDGFA to the organoid culture system supports matrix fibroblast gene expression. Other cellular compartments as a source of PDGFA such as the endothelium were not investigated in this study.61 62 Due to the simplified structure of organoids resulting in abnormal cell shape or by artefacts of the staining process caused by the presence of Matrigel, some immunohistochemical staining appears abnormal. For example, AGER is usually expressed in the membrane of AT1 cells, and while the staining of AGER in the organoids appears specific, it is expressed in the cytoplasm of some organoids. We used organoids generated with aged mouse lung epithelium as a negative control to set expression thresholds that detected specific protein expression to determine increased protein expression. Herein, mouse fibroblast–human epithelial cell organoids were used to generate organoids in which the fibroblasts were RA-treated prior to isolation. However, mouse human organoids may not fully recapitulate ageing in humans; therfore, further optimisation of the organoid systems in the future will allow for a more rigorous testing of the role of ageing in humans.
Taken together, our data suggest a model of paracrine signalling leading to alveolar repair in the aged lung, by which RA pretreatment of the fibroblast induces signalling to the epithelium that activates epithelial PDGFA secretion. Production of PDGFA by alveolar epithelial cells then activates pulmonary matrix fibroblast differentiation, which in return leads to restoration of alveolar differentiation after injury (figure 5).
Supplemental material
Data availability statement
Data are available in a public, open access repository. GEO: GSE157440.
Ethics statements
Patient consent for publication
Ethics approval
Ethics approval was obtained from Partners Institutional Review Board (2013P002332), Boston, Massachusetts, USA. Cincinnati Children’s Hospital Medical Center Institutional Review Board declared that donor tissue samples were Institutional Review exempt, in accordance with protocol 2013-3356.
Acknowledgments
The authors thank the generosity of the patients who contribute to the advancement of science.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Yanxu_Cincy
Correction notice This article has been corrected since it was published Online First. A minor modification has been made to the abstract.
Contributors JJG, JS, JG, MW, JJS, KEB, LPH, YX, AKTPJJG, JS and AKTP conceptualised the experiments; JJG, JG, MW and JJS performed the experiments; JJG, JG and MW analysed the data; JS performed the bioinformatic analysis; JJG, JS, KEB, LPH,YX and AKTP interpreted the results; KEB and LPH provided clinical and pathological expertise; JJG, JS and AKTP wrote the manuscript; and JG, MW, JJS, KEB, LPH and YX edited the manuscript.
Funding This work was supported by R01 HL131661 (to JG, YX, JS and AKTP); T32 HL007752 (to JJG); U01 HL122642 (to YX and AKTP), LungMAP1; U01 HL122638 (to YX and AKTP), LungMAP2; U01 HL134745 (to YX and AKTP), PCTC; K08 HL133603 (to KEB); and K23 HL132120 (to LPH). It was also supported by Translational Fibrosis Academic and Research Committee funding to CCHMC.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.