Article Text

Abstract
Introduction People living with HIV (PLWH) suffer from age-related comorbidities such as COPD. The processes responsible for reduced lung function in PLWH are largely unknown. We performed an epigenome-wide association study to investigate whether blood DNA methylation is associated with impaired lung function in PLWH.
Methods Using blood DNA methylation profiles from 161 PLWH, we tested the effect of methylation on FEV1, FEV1/FVC ratio and FEV1 decline over a median of 5 years. We evaluated the global methylation of PLWH with airflow obstruction by testing the differential methylation of transposable elements Alu and LINE-1, a well-described marker of epigenetic ageing.
Results Airflow obstruction as defined by a FEV1/FVC<0.70 was associated with 1393 differentially methylated positions (DMPs), while 4676 were associated with airflow obstruction based on the FEV1/FVC<lower limit of normal. These DMPs were enriched for biological pathways associated with chronic viral infections. The airflow obstruction group was globally hypomethylated compared with those without airflow obstruction. 103 and 7112 DMPs were associated with FEV1 and FEV1/FVC, respectively. No positions were associated with FEV1 decline.
Conclusion A large number of DMPs were associated with airflow obstruction and lung function in a unique cohort of PLWH. Airflow obstruction in even relatively young PLWH is associated with global hypomethylation, suggesting advanced epigenetic ageing compared with those with normal lung function. The disturbance of the epigenetic regulation of key genes not previously identified in non-HIV COPD cohorts could explain the unique risk of COPD in PLWH.
- COPD ÀÜ mechanisms
- immunodeficiency
- respiratory measurement
- viral infection
Data availability statement
Data are available upon reasonable request. Deidentified data are available upon reasonable request. The full methylation data will be deposited into a public repository in 2022 upon conclusion of the START study. Requests can be directed to Dr Janice Leung at janice.leung@hli.ubc.ca.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What explains the increased risk of COPD in patients living with HIV?
What is the bottom line?
Peripheral blood methylation disruptions are numerous in patients with both airflow obstruction and HIV infection.
Why read on?
Epigenetic disturbance related to chronic viral infections may hold clues to early disease-causing mechanisms in COPD.
Introduction
The progress in the treatment of HIV has led to an increase in life expectancy and a decrease in immunodeficiency syndrome-related conditions among people living with HIV (PLWH).1 Age-related comorbidities, though, have become common, including COPD,2 which is associated with increased mortality3 and significant respiratory symptom burden.4 Whether tobacco exposure, illicit drug use, repeated infections, or chronic inflammation are the key causes of this increased risk for COPD in PLWH is still unclear.
Lung function decline in PLWH was recently reported in a large, multinational cohort demonstrating that the timing of antiretroviral therapy initiation alone has no influence on the rate of decline.5 As in any population, the variability of lung function is likely the consequence of complex environmental and genetic factors, as well as their interaction6; however, the underlying molecular processes that explain these relationships in PLWH remain elusive. In this study, we explore the possibility that epigenetic alterations such as DNA methylation may influence lung function variability in PLWH. DNA methylation involves the addition of a methyl group to a cytosine base located next to guanine base (CpG site). The methylation of CpG sites in regulatory elements (ie, promoter regions) often results in decreased gene expression7 and can potentially affect other traits. Environmental factors such as tobacco use and chronic diseases or infections as well as the natural ageing process can all influence DNA methylation. Age-related diseases, for instance, are characterised by genome-wide hypomethylation.8 The methylation of ubiquitous transposable elements like Alu and LINE-1 is used as markers for global methylation and is thought to play key roles in age-related genomic instability,9 which may lead to tumorigenesis and senescence.
Previous efforts to understand the effect of DNA methylation on lung function have focused mainly on non-HIV cohorts.10 11 Evidence from these studies suggests, however, that methylation may play an important role in lung function and the aetiology of COPD.12 For this study, we conducted an epigenome-wide association analysis to investigate the relationship of blood DNA methylation with lung function of PLWH. We hypothesised that PLWH with airflow obstruction have a distinct methylation pattern when compared with those with normal lung function. We also hypothesised that differential DNA methylation is associated with lung function decline in PLWH.
Methods
Study cohort
The study cohort consisted of 161 PLWH over the age of 40 years who were enroled in the genomic and the pulmonary substudies of the Strategic Timing of Antiretroviral Therapy (START, Clinicaltrials.gov NCT00867048) trial, which has been previously described.13 14 Briefly, this was a multicentre, international, randomised controlled trial designed to compare immediate versus deferred initiation of antiretroviral therapy. The START cohort included adult PLWH with CD4 T cell counts >500 cells/mm3 and who had not yet been exposed to antiretroviral therapy.5
Lung function and filtering criteria
All participants underwent spirometry testing yearly for up to 6 years, according to methods previously described.5 Participants with three or more spirometric tests were retained for the analysis on FEV1 decline. In total, 152 participants were retained for FEV1 decline analysis, while all 161 subjects were included in cross-sectional analyses (online supplemental figure S1). Participants were characterised as having airflow obstruction if the FEV1/FVC ratio was <0.70. In addition, airflow obstruction was also assessed based on FEV1/FVC ratio <lower limit of normal (LLN), according to the Global Lung Function Initiative 2012 normative equations.15
Supplemental material
DNA methylation profiling and quality control
Participants had a whole blood sample drawn at study entry. The DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) was used to extract DNA from this sample. Unmethylated cytosine residues present in the DNA extract were converted to uracil using the EZ DNA Methylation Kit (Zymo, Irvine, California, USA). DNA methylation profiles were obtained using the Illumina Infinium MethylationEPIC BeadChip microarray which interrogates 863 904 CpG sites and covers 95% of all genes and 95% of CpG islands.16 The ratio of the methylated probe intensity to the overall intensity (β value) was calculated for each CpG and ranged from 0 (all unmethylated) to 1 (all methylated) and then transformed to M-values (log2 ratio of the intensity of the methylated probe to unmethylated CpG probe). CpG probes were filtered based on the detection quality and probes with a detection p>1e−10 were excluded from downstream analyses. In addition, non-CpG, XY-linked, single nucleotide polymorphism (SNP) and cross-hybridisation probes were also removed (839 418 CpGs were retained). Lastly, background correction, normalisation and batch correction were performed using the normal–exponential out-of-band,17 mixture quantile normalisation18 and ComBat19 methods, respectively.
Alu and LINE-1 imputation
Global methylation status can be inferred from the methylation of repetitive and transposable elements along the genome, of which Alu and LINE-1 are among the most abundant.20 Hypomethylation of these sites is associated with ageing as well as with worse lung function in non-PLWH study cohorts.21 Alu and LINE-1 sites were imputed using the machine learning tool repetitive element methylation prediction.22
Statistical analysis
The cell type proportion of each sample was estimated using the deconvolution method of Houseman et al 23 which provides the proportion of CD4 T cells, CD8 T cells, natural killer cells, monocytes, granulocytes (neutrophils+eosinophils) and B cells. The EPISTRUCTURE software24 was used to infer the ancestry. This software calculates the principal components using CpGs that are highly correlated with SNPs, capturing the genetic variation within a population. Additional covariates were chosen based on the algorithm outlined by Lee et al. 25 To identify differentially methylated positions (DMPs) between PLWH with airflow obstruction and with normal lung function, we performed an epigenome-wide association study using a robust linear model (rlm) implemented in the MASS R package (M-estimation),26 and adjusted as follows:

The model presented above was also used to interrogate Alu and LINE-1 methylation sites.
To investigate the effect of methylation over cross-sectional FEV1, and FEV1/FVC (at baseline visit) in PLWH we also used rlm adjusted for the following covariates:

The effect of methylation over FEV1 decline over the course of 6 years was studied using a random coefficient model (lme) implemented in the nlme R package27; both a random intercept term and a random slope term were included and the model was adjusted for the following covariates:
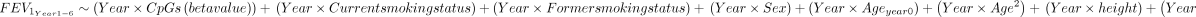
The DMPs for each model were defined at a false discovery rate (FDR)<0.10. The R package DMRcate28 was used to identify differentially methylated regions (DMRs), defined with at least three CpGs. The R package clusterProfiler was used to identify Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that were significantly (FDR<0.10) enriched by genes that corresponded to DMPs. The FEV1 decline analysis was executed first for all PLWH, then additionally separately by each ethnic group.
Results
Description of the study cohort
The baseline demographic characteristics of the study cohort grouped by airflow obstruction status based on the fixed ratio criteria are shown in table 1. Demographic data grouped by airflow obstruction based on the LLN criteria are shown in online supplemental table S1. Fifteen individuals met both airflow obstruction criteria, whereas five met criteria only for FEV1/FVC<0.70 and one met criteria only for FEV1/FVC<LLN. Based on Mann-Whitney U tests, the airflow obstruction group had a larger proportion of smokers and lower body mass index (p<0.05) than the normal lung function group, irrespective of the criteria used for airflow obstruction characterisation. There was no significant difference in CD4 T cell counts and HIV RNA viral load between the groups.
Supplemental material
Summary of the study cohort by airflow obstruction group (FEV1/FVC<0.70 criteria)*
Airflow obstruction in PLWH is associated with methylation
We identified 1392 DMPs (online supplemental table S2) and 2 DMRs (online supplemental table S3) that were associated with airflow obstruction (based on the FEV1/FVC ratio <0.70 criteria). Of these, 846 DMPs were hypermethylated in individuals with airflow obstruction, while 546 were hypomethylated. Twenty-eight per cent of DMPs were located in CpG Islands and 46% in genomic region of low CpG density (Open Sea), in addition 21% of DMPs were within promoter regions (online supplemental table S4).
Based on the criteria of FEV1/FVC ratio <LLN, 4675 DMPs and 9 DMRs were identified (online supplemental tables S2 and S3). We found 2843 hypomethylated DMPs in PLWH experiencing airflow obstruction, while 1832 were hypermethylated. The majority of DMPs were in the Open Sea region (61%), while 14% are in CpG Islands and 11% in promoter regions (online supplemental table S4). Moreover, there were 745 DMPs that overlapped between the analyses by FEV1/FVC ratio <LLN and FEV1/FVC ratio <0.70 criteria.
Figure 1 shows the range of statistical significance and methylation beta difference for DMPs distinguishing those with and without airflow obstruction. The most significant DMPs correspond to the genes HK2, HBEGF, TAPBP, MAD1L1, GPR153, VGLL4 and ADCY7 (table 2). The DMPs for airflow obstruction (criteria: FEV1/FVC<0.70) enriched multiple KEGG pathways including ‘Small cell lung cancer’, ‘Hepatitis B’, ‘Epstein-Bar virus infection’ and ‘Human Papillomavirus infection’. The top 10 pathways are shown in figure 1C. Only one biological pathway, cAMP signalling pathway, was enriched by DMPs for the FEV1/FVC<LLN criteria.
Most significant differentially methylated positions (DMPs) for airway obstruction in people living with HIV

Differentially methylated points (DMPs) for airflow obstruction and top 10 biologic-enriched pathways for airflow obstruction (C). The x-axis on A and B represents the effect size difference of the DMPs between subjects with and without airflow obstruction (reference group: no airflow obstruction). The y-axis on figures A and B represents the level of statistical significance for each DMPs. Airflow obstruction was defined as (A) FEV1/FVC ratio was <0.70 and (B) FEV1/FVC <lower limit of normal. The red and blue colours represent hypomethylation and hypermethylation, respectively. The dashed horizontal line in A and B represents the −log10 p value that corresponds to the false discovery rate (FDR)<0.10. The axis in C represents the enrichment level of significance (×) for each biological pathway (y). The size of the circles inside the figure represents the number of overlapping genes in the pathways and the genes characterised by DMPs. The colour green represents significant enrichment based on the FDR<0.1. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Global methylation: Alu and LINE-1 sites
We investigated the overall differences in methylation between those with and without airflow obstruction using the Alu and LINE-1 CpGs as markers of global methylation. Results show that of 122 differentially methylated Alu sites and 13 differentially methylated LINE-1 sites, 117 and 12, respectively, were hypomethylated in those with a FEV1/FVC ratio <70% (online supplemental table S5). Moreover, of 781 differentially methylated Alu sites and 105 LINE-1 sites, 768 and 101, respectively, were hypomethylated in those with a FEV1/FVC ratio <LLN (online supplemental table S5 and figure 2).

{kind=link}
{kind=link}
Alu and LINE-1 differentially methylated sites in people living with HIV with airflow obstruction. (A) and (B) Differentially methylated points (DMPs) in transposable element Alu between subjects with and without airflow obstruction (reference group: without airflow obstruction). (C) and (D) DMPs in transposable element LINE-1 between subjects with and without airflow obstruction (reference group: without airflow obstruction). The x-axis on the plots represents the effect size difference of the DMPs between subjects with and without airflow obstruction. The y-axis on the plots represents the level of significance for each DMPs. Airflow obstruction was defined as FEV1/FVC ratio was <0.70 (A and C) and FEV1/FVC <lower limit of normal (B and D). The dashed horizontal line inside the plots represents the −log10 p value that corresponds to the false discovery rate<0.10.
Cross-sectional lung function and methylation
We assessed blood methylation and lung function as a continuous measure in PLWH and identified 103 DMPs and 9 DMRs associated with FEV1 (online supplemental tables S2 and S3). The absolute effect of a 1% change in methylation of the DMPs for FEV1 was on average 110.69 mL and ranged from 1.71 mL to 850.63 mL (online supplemental figure S2). In total, 7112 DMPs and 888 DMRs were identified as having an association with FEV1/FVC (online supplemental tables S2 and S3). The average effect size of a 1% change in methylation of DMPs on FEV1/FVC was 1.67%, and the minimum and maximum were 0.10% and 27.03%, respectively. While the DMPs for FEV1 did not enrich any KEGG pathways, online supplemental figure S3 shows that DMPs for FEV1/FVC enriched multiple KEGG pathways, the most significant of which was inflammatory mediator regulation of transient receptor potential channels.
The majority of the DMPs were located in regions with low CpG density (Open Sea and Shelf) (online supplemental table S4). Approximately 45% of FEV1 DMPs were located within regions with high density (CpG Island) or close to CpG Islands (Shores), while 13.97% and 8% of FEV1/FVC ratio DMPs were located in CpG Islands and Shores, respectively. In addition, 4.8% and 11.14% of DMPs influencing FEV1 and FEV1/FVC, respectively, are located within known promoter regions.
Table 3 shows the seven genes corresponding to the top DMPs associated with lung function, which include MACROD1, DPF3, CACNA1G, PADI4, DLEC1, FERMT3 and ELANE. Overall hypermethylation of the top DMPs was associated with decreased FEV1. Moreover, 78% of all DMPs for FEV1 also have negative effects. Similar results were found for the FEV1/FVC ratio, where 86% of the DMPs had effect sizes with a negative direction. The top DMR associated with FEV1 included 7 CpGs and corresponding to CTHRC1, while the top DMR for FEV1/FVC included 27 CpGs and correspond to CTSZ (online supplemental tables S3).
Most significant differentially methylated positions for baseline FEV1 and FEV1/FVC
Lung function decline
Over a median of 5 years, PLWH had a small decline in FEV1 (online supplemental figure S4). Overall FEV1 declined on average by 20 mL/year; however, the decline was not statistically significant (p=0.43, 95% CI −187.04 to 430.19). Caucasian PLWH showed an overall FEV1 decline, but this was not statistically significant either (32 mL/year, p=0.33, 95% CI −205.06 to 586.28). There were no DMPs associated with FEV1 decline in the overall cohort. In order to remove the noise in FEV1 decline that was due to the variability between ethnic groups, we analysed Africans (n=31), Caucasians (n=97) and Hispanics (n=20) separately; however, only the Caucasians yielded significant results. We found 53 DMPs and 4 DMRs (online supplemental tables S2 and S3) that were significantly associated with FEV1 decline in Caucasian PLWH. The top five DMPs are shown in table 4; two of them, cg09595479 and cg08625260, were located within a CpG Island and corresponded to PRPH and IRS2, respectively.
Most significant differentially methylated positions (DMPs) for FEV1 decline in Caucasian people living with HIV
Discussion
This study is the first epigenome-wide association analysis on airflow obstruction and lung function in a multiethnic cohort of PLWH. Previous research on DNA methylation and lung function has focused on general or COPD-specific populations.12 29 Because DNA methylation can be altered by environmental factors including chronic infections, these past results may not reflect the relationship between methylation and lung function in the HIV-specific context. Our study revealed that PLWH with airflow obstruction have a distinct blood DNA methylation profile compared with PLWH with normal lung function, and that airflow obstruction is linked with global hypomethylation in HIV. Furthermore, our results indicate that although DNA methylation is associated with cross-sectional lung function, there was minimal influence on lung function decline.
Most methylated CpGs in the genome are located in CpG-rich sequences of the transposable elements Alu and LINE-1; therefore, CpG sites within these elements are often used as markers for global methylation. Hypomethylation along these elements has also been used as a marker for ageing9 and has also been associated with lower lung function in healthy older men.21 Our results on Alu and LINE-1 show that those PLWH with airflow obstruction have greater hypomethylation when compared with those with normal lung function. This is despite the fact that subjects with airflow obstruction in our cohort still had relatively mild decrements in FEV1 and were relatively young. These results suggest that the process of airflow obstruction in PLWH may reflect an advanced ageing process, concurrent with the observations of accelerated ageing and comorbid age-related conditions in HIV.30 The biological implications of global hypomethylation and lung obstruction need further investigation, however given that global hypomethylation can potentially lead to overexpression of genes and activation of transposable elements and thus promote tumourigenesis in the lung,31 the interplay between smoking, airflow obstruction and lung cancer may be mediated by this specific methylation process. As PLWH are at higher risk of developing lung cancer,32 this hypothesis should be explored further.
The top pathways enriched by DMPs for airflow obstruction included the Hepatitis B, Epstein-Barr virus and Human Papillomavirus pathways, which raises the intriguing possibility that concurrent viral infections in PLWH may be drivers of airflow obstruction. PLWH are known to be at higher risk of coinfection with these particular viruses,33–35 although to our knowledge the association of these chronic infections with COPD from an epidemiologic standpoint has not yet been reported. Another top pathway enriched by DMPs, the cAMP signalling pathway, is related to small airway remodelling in COPD, and therapeutic compounds that target proteins in this pathway such as roflumilast have been used to treat COPD.36 Furthermore, differentially methylated genes in the small airways of patients with COPD have been also found to be enriched in the cAMP signalling pathway.29 While more research is needed to validate that the differential methylated genes could alter enriched biologic pathways, no previous research has linked these pathways at the DNA methylation level with airflow obstruction in PLWH.
Because of the uniqueness of our study cohort, a multiethnic group of PLWH who were ART naïve at study entry, we could not replicate this analysis. However, there is a modest overlap between genes identified in our study with previous work looking at methylation in COPD populations (online supplemental figure S5 and table S2). In accordance with previous findings on the small airway methylation profiles of patients with COPD,29 we identified a large number of DMPs associated with airflow obstruction in PLWH . One of our discovered DMPs (cg13071306) corresponds to a gene previously described in airways diseases, GPR153. The function of GPR153 is poorly understood; however, this gene belongs to a rhodopsin family of G protein-coupled receptors (GPCRs), which are mediators of airway smooth muscle contraction and increased airway resistance. GPCRs, for example, are frequently dysregulated in asthma.37 In addition, one of our top hits for airflow obstruction (cg01175605) is located in an exon of HK2, which has previously been linked to COPD and lung cancer. HK2 is a hexokinase predominantly localised to the mitochondrial membrane as part of the glucose metabolism pathway, but has also been reported to be expressed in the lung.38 Specifically, the CpG site cg18638581 in the promoter region of HK2 was associated with COPD, FEV1 and FEV1/FVC in a previously reported COPD cohort.39 This effect was independent of tobacco use. Previous work has demonstrated as well that HK2 is upregulated in non-small cell lung cancer.40 Possible regulation of HK2 expression may occur through epigenetic changes to influence the development of COPD and lung cancer.
While this study provides novel findings, it also has several limitations. First, our study cohort was restricted to PLWH over 40 years of age, with a detectable viral load and CD4 T cell count >500 cells/mm3, and who were not at the time of study entry on antiretroviral therapy. Whether these results apply to PLWH who have been on antiretroviral therapy for many years and have achieved viral suppression cannot be ascertained here. Second, the FEV1 decline analysis suggested that decline is not likely to be affected by methylation changes; however it is also possible that our analysis of FEV1 decline was simply underpowered. The proportion of our cohort meeting criteria for airflow obstruction was small and analyses of methylation in cohorts with a greater fraction of patients with COPD should be performed in the future. It is possible that some of the effects identified by our study also apply to non-HIV cohort; however this was outside the scope of our study. Third, the direction of effect, whether DNA methylation disruptions influence the progression of airflow obstruction or conversely whether airflow obstruction alters DNA methylation profiles cannot be ascertained by these data. Further study in cohorts with longitudinal DNA methylation profiling would be essential to solving this problem. Finally, because of the pressing need to extrapolate findings to diverse populations of PLWH, we included multiple ethnic groups in our analysis while controlling for population structure to the best of our abilities. However, since some methylation sites are specific to certain ethnicities, and would only be identified in homogenous populations, future efforts should focus on increasing the sample size of underrepresented minority groups. Despite these limitations, we have identified for the first time linkages between lung function, airflow obstruction and methylation in a unique cohort of PLWH. Epigenetic disruptions at key genes may hold clues to the increased risk of chronic lung diseases in this population.
Data availability statement
Data are available upon reasonable request. Deidentified data are available upon reasonable request. The full methylation data will be deposited into a public repository in 2022 upon conclusion of the START study. Requests can be directed to Dr Janice Leung at janice.leung@hli.ubc.ca.
Ethics statements
Ethics approval
The study was conducted under the University of British Columbia Ethics Board Approval Number H15-02166.
Acknowledgments
The authors thank the study participants who were involved in the START trial (see reference [13] for the complete list of START investigators, and [14] for the complete list of START Pulmonary Sub study investigators). They used computational resources from Compute Canada and WestGrid to conduct this research.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AIHC, CXY, MO, SFPM, DDS, KK, JL: study design and conception, interpretation of data, manuscript drafting and editing. JY: data acquisition, manuscript drafting and editing. JM, DL, RN, FH, HK, ND: data acquisition, manuscript editing. LM: data acquisition, interpretation of data, manuscript editing. MK: study design, interpretation of data, manuscript editing.
Funding AIHC is supported by MITACS accelerate. This study was supported by the Canadian Institutes of Health Research (F15-00958). JML is supported by the Michael Smith Foundation for Health Research and the Canadian Institutes of Health Research. The START Pulmonary Substudy was supported by the National Heart Lung and Blood Institute [R01 HL096453]; the parent START trial was primarily supported by the National Institute of Allergy and Infectious Diseases Division of AIDS [UM1 AI068641 and UM AI120197] with additional support from the German Ministry of Education and Research, the European AIDS Treatment Network (NEAT), the Australian National Health and Medical Research Council, and the UK Medical Research Council and National Institute for Health Research. The Veterans Health Administration Office of Research and Development also provided protected research time in support of this study. The University of Minnesota served as sponsor of the study.
Disclaimer None of the funders nor sponsor had any input regarding the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.