Article Text

Abstract
Thrombotic events that frequently occur in COVID-19 are predominantly venous thromboemboli (VTE) and are associated with increasing disease severity and worse clinical outcomes. Distinctive microvascular abnormalities in COVID-19 include endothelial inflammation, disruption of intercellular junctions and microthrombi formation. A distinct COVID-19-associated coagulopathy along with increased cytokines and activation of platelets, endothelium and complement occur in COVID-19, which is more frequent with worsening disease severity. This proinflammatory milieu may result in immunothrombosis, a host defence mechanism that can become dysregulated, leading to excess formation of immunologically mediated thrombi which predominantly affect the microvasculature. The haemostatic and immune systems are intricately linked, and multifactorial processes are likely to contribute to VTE and immunothrombosis in COVID-19. This state-of-the-art review will explore the pathobiological mechanisms of immunothrombosis and VTE in COVID-19 focusing on: COVID-19-associated coagulopathy, pathology, endothelial dysfunction and haemostasis, the immune system and thrombosis, genetic associations and additional thrombotic mechanisms. An understanding of the complex interplay between these processes is necessary for developing and assessing how new treatments affect VTE and immunothrombosis in COVID-19.
- pulmonary embolism
- innate immunity
- viral infection
- cytokine biology
- respiratory infection
This article is made freely available for use in accordance with BMJ’s website terms and conditions for the duration of the covid-19 pandemic or until otherwise determined by BMJ. You may use, download and print the article for any lawful, non-commercial purpose (including text and data mining) provided that all copyright notices and trade marks are retained.
https://bmj.com/coronavirus/usageStatistics from Altmetric.com
Introduction
COVID-19 is caused by the novel SARS-CoV-2 that has disseminated in a global pandemic. SARS-CoV-2 is a single-stranded RNA virus, of the genus betacoronavirus, that enters cells via ACE 2 (ACE2) receptors.1 SARS-CoV-2 has homology to SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV), which caused the 2002/3 SARS and 2012 MERS outbreaks, respectively.1–3 COVID-19 pneumonia can cause fever, cough and dyspnoea with approximately 15% of cases being severe and 5% requiring intensive care support for acute respiratory distress syndrome (ARDS) or multiorgan failure.4 Demographics and comorbidities that are associated with COVID-19 include older age, male sex, ethnicity, diabetes, systemic hypertension and chronic cardiorespiratory disease.5 6
Thrombotic events occur in up to one-third of patients with COVID-19, which are predominately pulmonary emboli, and are associated with more severe disease and increased mortality.7 8 However, studies are heterogenous and incidence varies by cohort composition (eg, disease severity, definition of thrombotic events), investigations performed and the use of thromboprophylaxis.9 Venous thromboembolism (VTE) incidence is high in other viral diseases including SARS-CoV-1 and H1N1; however, a direct comparison with COVID-19 is challenging due to varied cohorts and methodologies.10 11 VTE rates are higher in severe COVID-19 than matched groups with ARDS, suggesting the high incidence is due to mechanisms in addition to VTE risk factors in hospitalised patients (eg, immobility and severe illness).12 Furthermore, VTE may be under-recognised in COVID-19 as the incidence increases when screening investigations are performed; however, this may also apply to other diseases.13 Smaller pulmonary (micro)thrombosis in COVID-19 may represent in situ immunothrombosis, a process initiated by the innate immune system that involves cross-talk with haemostasis.8 14 15 There is current uncertainty about whether COVID-19-associated thrombotic events are due to conventional VTE, immunothrombosis or a combination, which has important implications for diagnostic and management strategies.
We aim to review the pathobiological mechanisms of immunothrombosis and VTE in COVID-19 focusing on: COVID-19-associated coagulopathy (CAC), pathology, endothelial dysfunction and haemostasis, the immune system and thrombosis, genetic associations and additional thrombotic mechanisms. A literature search (MEDLINE) was performed in June 2020 using the following search terms (including variations and acronyms): “Covid-19”, “venous thromboembolism”, “immunothrombosis”, “coagulopathy”, “endothelial dysfunction”, “platelet”, “cytokine” and “complement”.
COVID-19-associated coagulopathy
Patients with COVID-19 can have mild thrombocytopenia, mildly prolonged prothrombin time, increased fibrinogen and raised D-dimer (table 1), all of which are more pronounced as disease severity increases.16 17 This pattern of CAC shares features with sepsis-induced coagulopathy (SIC) and disseminated intravascular coagulation (DIC), but is a distinct entity.18 DIC and SIC can occur in COVID-19, but are less common when validated diagnostic criteria are applied.18 Similar CAC findings have also been reported in SARS-CoV-1 infections.10
Blood markers of coagulation, fibrinolysis and inflammation in COVID-19
D-dimer is a fibrin degradation product that is sensitive at detecting fibrinolysis of intravascular thrombus (ie, VTE) but lacks specificity and can be raised in inflammation and other diseases.19 A marked increase in D-dimer can occur in COVID-19 and has been independently associated with mortality.20 21 Raised D-dimer may be related to COVID-19 acute lung injury and produced by the breakdown of intra-alveolar fibrin, which is deposited in ARDS.22 23
Additional markers of coagulation and inflammation can also be abnormal in COVID-19 including ferritin, von Willebrand Factor (VWF), C reactive protein (CRP), complement and cytokines (table 1). This suggests a complex interaction between the haemostatic and immune systems that may contribute to a prothrombotic phenotype that is discussed in this review.
COVID-19 pathology
SARS-CoV-2 has a predilection for the respiratory tract, gaining cellular entry via the ACE2 receptor that is expressed on the surface of airway epithelial cells.24 25 Pathological changes in COVID-19 include diffuse alveolar damage, activation of type II pneumocytes, hyaline membrane formation and fibrin deposition; changes consistent with ARDS.26 27 Distinctive pulmonary microvascular abnormalities occur in COVID-19 that include intravascular fibrin deposition, perivascular monocyte infiltration, angiogenesis and microthrombi formation.26 27 Pulmonary endothelial cell inflammation, membrane disruption and damage are prominent features that may result from direct viral effects, consistent with endothelial ACE2 receptor expression, or indirect host inflammatory effects.23 28 Importantly, the microvascular changes in COVID-19 are more pronounced than in H1N1-infected lungs, suggesting disease-specific effects rather than epiphenomenon of ARDS or viral pneumonia.23 VTE occurs in up to 50% of COVID-19 autopsy series and the frequent occurrence of DVT suggests embolic complications in addition to in situ microvascular immunothrombosis.26 29 The ACE2 receptor is widely expressed by different cells and SARS-CoV-2 has been detected in the kidneys, liver, heart and brain, which may account for extrapulmonary thrombotic complications along with the ubiquitous presence of endothelium in different organs.7 25 30
Pulmonary thrombosis in COVID-19 may represent conventional PEs or immunothrombosis (particularly for smaller clots); however, there is no current differentiating diagnostic strategy. The pathological changes from autopsy series suggest a combination of these two processes occurs in COVID-19, although this may only apply to severe disease.
Endothelial dysfunction and haemostasis
The endothelium is a monocellular layer lining blood vessels with functions that include providing a mechanical barrier between circulating blood and the basement membrane, controlling vascular tone and immunomodulation.31 Endothelial dysfunction involves endothelial activation and reduced endothelium-dependent vasodilation, which results in a proinflammatory, procoagulant and proliferative state.32 COVID-19 clinical outcomes are worse in patients with diseases associated with endothelial dysfunction (eg, systemic hypertension, diabetes and obesity) and evidence of endothelial dysfunction is present in COVID-19 autopsy series.23 33 The mechanism(s) for endothelial dysfunction could occur via direct SARS-CoV-2 invasion of endothelial cells or indirect inflammatory effects.23 34 Binding of the SARS-CoV-2 spike protein to the ACE2 receptor is facilitated by host serine protease TMPRSS2 priming, followed by viral endocytosis and replication.24 34 Subsequent endothelial damage and viral release triggers a marked immune response that could cause additional endothelial dysfunction (see ‘The immune system and thrombosis’ section).
Haemostasis overview
A prothrombotic state occurs in COVID-19 that could be a consequence of increased coagulation, decreased fibrinolysis and immune effects. Coagulation involves a complex biological cascade and is a component of haemostasis along with vascular spasm and platelet activation. Endothelial damage and disruption of intercellular junctions in COVID-19 exposes the subendothelial matrix containing tissue factor (TF) and collagen.23 35 This activates the coagulation cascade and results in thrombin generation and conversion of fibrinogen to fibrin which, together with platelet aggregates, forms blood clots (figure 1).35 Mild prolongation of prothrombin time in CAC (particularly in severe disease) could signify activation of the TF (extrinsic) pathway (figure 1). TF is a subendothelial transmembrane protein and FVII/FVIIa cofactor that potently activates the coagulation cascade.36 37 In COVID-19, TF expression on macrophages and platelets could be induced by inflammatory cytokines.36 Furthermore, tissue factor pathway inhibitor (TFPI), which inhibits the TF pathway, could be impaired by inflammation in COVID-19 leading to further coagulation.38 Endogenous anticoagulant levels (α2-antiplasmin, protein C/S, antithrombin) are normal in COVID-19, which is further evidence that CAC is distinct from DIC.39 Markers of endothelial activation (VWF, FVIII, P-selectin) are increased in COVID-19, and raised soluble thrombomodulin (an endothelial glycoprotein) together with VWF are associated with worse clinical outcomes.39 Importantly, the presence of endothelial activation and haemostatic abnormalities in intensive care unit (ICU) and non-ICU patients with COVID-19 suggests these processes are important in disease pathophysiology and not only an epiphenomenon of ARDS (figure 2).39 40

Clotting cascade abnormalities in COVID-19. The coagulation cascade is initiated by exposure to prothrombotic proteins from the subendothelium or following expression of tissue factor, referred to as the intrinsic pathway and extrinsic pathways, respectively. Clinical investigation of the intrinsic and extrinsic pathways can be measured by activated partial thromboplastin time (aPTT) and prothrombin time (PT), respectively. A series of sequential cleavages occur, whereby proteolytic coagulation factors convert circulating, inactive factors into their active form. Both pathways reach a common pathway, by which activated factor X cleaves prothrombin to form thrombin. Thrombin cleaves fibrinogen to give rise to fibrin strands, which rapidly polymerise to stabilise platelet aggregates and form a thrombus. Fibrin can be degraded by plasmin; tissue plasminogen activator (tPA) facilitates the cleavage of plasminogen to give rise to plasmin. Components of the clotting cascade that are abnormal or putatively associated with COVID-19, particularly severe disease, are shown in shaded symbols. In COVID-19, tissue factor pathway inhibitor (TFPI), factor VIII, fibrinogen, plasminogen-activator inhibitor-1 (PAI-1) and fibrin degradation products (FDPs) have been shown to be elevated. A prolonged PT has also been reported. Studies have shown reduced platelet numbers and levels of tPA in COVID-19. Elevated parameters are indicated with a ↑ symbol, decreased parameters with a ↓ symbol.
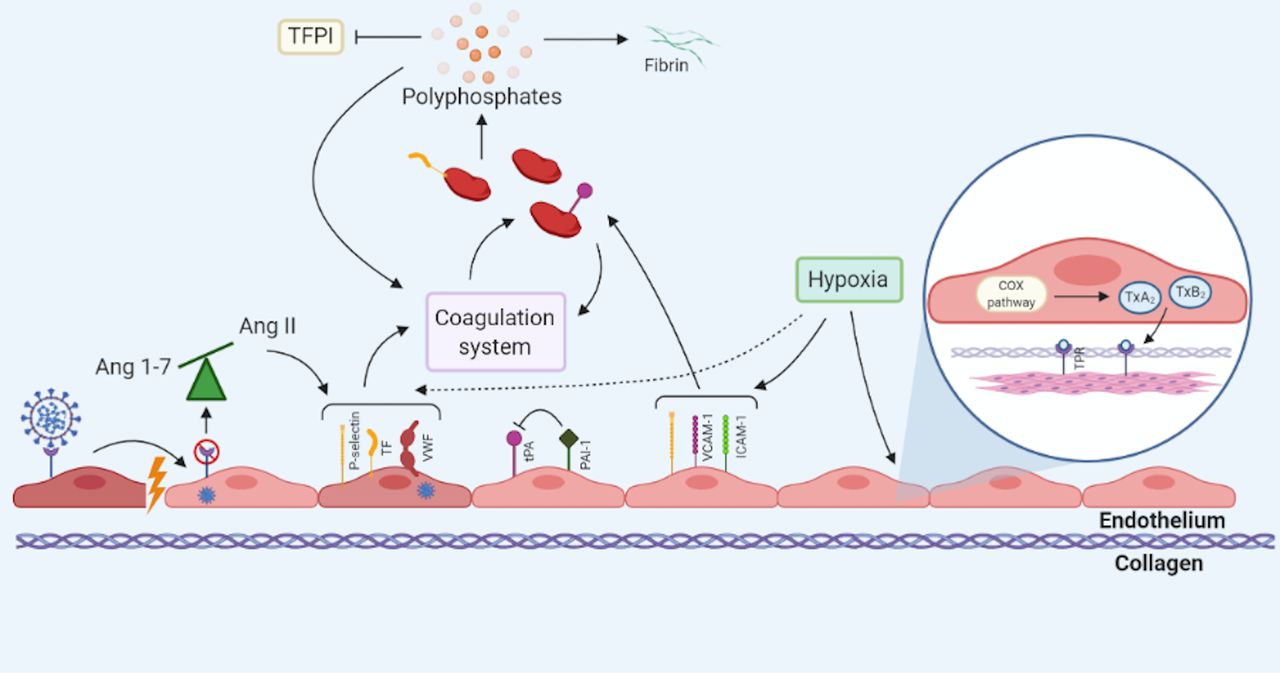
Endothelial dysfunction in COVID-19. Endothelial dysfunction in COVID-19 may occur through multiple mechanisms and precipitating factors. Direct invasion of SARS-CoV-2 into endothelial cells causes cellular damage that disturbs intercellular junctions and exposes prothrombotic subendothelial collagen. Internalisation of the ACE2 receptor causes an imbalance of Ang1-7 and AngII, in favour of the latter. Accumulation of AngII promotes the endothelial expression of P-selectin, tissue factor (TF) and von Willebrand factor (VWF). Intracellular viral replication within endothelial cells results in their activation and expression of an array of prothrombotic proteins. Expression of these prothrombotic proteins activates the extrinsic coagulation cascade. Simultaneous recruitment of platelets to the site of endothelial injury further contributes to hypercoagulability. Polyphosphates are secreted by activated platelets and promote the activation of coagulation factors. Polyphosphates inhibit tissue factor pathway inhibitor (TFPI) and encourage fibrin polymerisation. Local hypoxia exacerbates the prothrombotic phenotype, through induction of P-selectin, TF and VWF expression on the endothelial surface. Hypoxia-induced activation of the cyclooxygenase (COX) pathway releases thromboxanes A2 and B2 (TxA2 and TxB2, respectively). TxA2 and TxB2 bind to thromboxane prostanoid receptors (TPRs) present on smooth muscle cells, resulting in vasoconstriction. ICAM-1, intercellular adhesion molecule-1; PAI-1, plasminogen-activator inhibitor-1; tPA, tissue plasminogen activator; VCAM-1, vascular cell adhesion molecule-1.
Fibrinolysis
Reduced fibrinolysis has been described in severe COVID-19 and increased VTE occurs in patients with more severe abnormalities of clot dissolution.16 The combination of raised D-dimer (a marker of fibrinolysis) and evidence of apparent hypofibrinolysis has been proposed to either be a consequence of differences between systemic and local effects, or due to the fibrinolytic system becoming overwhelmed.41 42 The fibrinolytic inhibitor plasminogen activator inhibitor 1 (PAI-1) is increased in COVID-19, SARS-CoV-1 infection and other causes of ARDS where hypofibrinolysis and fibrin deposition are hallmark features.39 43 Inflammation promotes PAI-1 release from endothelial cells, which suppresses urokinase-plasminogen activator and tissue-type plasminogen activator (tPA) from converting plasminogen to plasmin, which ultimately leads to reduced fibrin degradation.43 PAI-I is increased in ICU and non-ICU patients with COVID-19, suggesting a role in disease pathobiology and progression that is not only related to ARDS.39
Platelets
Activated endothelial cells express a number of proteins including P-selectin, a cell adhesion molecule that enables the recruitment of platelets and leucocytes, which have a pivotal role in haemostasis and thrombosis.44 Disruption of the endothelial layer exposes the collagen containing subendothelial matrix and also results in platelet activation and recruitment.45 Subsequent platelet degranulation and aggregation produces a platelet plug that functions as an adhesion site for coagulation factors.45 Activated platelets secrete a range of bioactive molecules (eg, ADP, polyphosphates, coagulation factors) and immunological mediators (eg, complement factors) that cause further platelet activation and amplification of the immune system via positive feedback mechanisms contributing to haemostasis.45
Platelet counts are normal or mildly reduced in COVID-19, unless there is concurrent DIC which is uncommon.18 However, marked platelet activation occurs with rapid aggregation and increased platelet-leucocyte aggregates that are more pronounced in severe COVID-19.46 47 Markers of platelet activation (eg, P-selectin, soluble CD40L) are increased in COVID-19 and P-selectin can induce monocyte TF expression, leading to a procoagulant phenotype.39 47 The glycoprotein VWF produced by activated endothelial cells, platelets or exposed subendothelium mediates platelet adhesion and aggregation.48 VWF is markedly increased in COVID-19, which could signify a propensity for platelet plug formation and thrombosis.39 Platelets have an important function in the innate immune system and activated platelets release complement (C3), which may contribute to COVID-19 immunothrombosis (see ‘The immune system and thrombosis’ section).49
Hypoxia
Hypoxia occurs in moderate-to-severe COVID-19 and this can lead to endothelial dysfunction and hypercoagulability.50 51 Upregulation of endothelial P-selectin and adhesion molecules (eg, intercellular adhesion molecule-1 (ICAM-1)) in hypoxia results in platelet and leucocyte recruitment.52 Monocytes bind to activated endothelial cells through the P-selectin glycoprotein ligand-1, and further express prothrombotic factors such as TF.53
Hypoxia-induced factors (HIFs) are transcription factors expressed by endothelial and immune cells in response to hypoxaemia.54 HIFs promote thrombosis by increasing endothelial release of PAI-1 and inflammatory cytokines (eg, tumour necrosis factor (TNF), interleukin (IL)-2), while downregulating thrombomodulin.51 52 Additionally, HIF activity can initiate the immune system; a hypoxic environment can cause release of damage-associated molecular patterns (DAMPs), that potently trigger an immune response (see ‘The immune system and thrombosis’ section). In macrophages, HIFs promote their activation and local aggregation, along with driving the expression of proinflammatory cytokines including IL-6 and TNF-ɑ.54 HIF-1α could enhance complement-mediated endothelial damage in COVID-19 by decreasing the expression of the complement regulator CD55.55 The cumulative effects of hypoxia are a likely contributor to dysregulated haemostasis and disruption of vascular tone in COVID-19.
Vasoconstriction
Loss of vascular tone is a feature of endothelial dysfunction and, when culminating in vasoconstriction, can have prothrombotic consequences. A number of hypoxia-dependent pathways can drive this process. Hypoxia-induced expression of adhesion molecules, namely P-selectin, E-selectin, ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1) disrupts the endothelium. Subsequent increase in microvascular permeability exposes the subendothelial matrix, rapidly triggering thrombosis.52 Alveolar and tissue hypoxia in severe COVID-19 may initiate the cyclooxygenase (COX) pathway in endothelial cells; binding of COX-induced thromboxanes A2 and B2 to thromboxane prostanoid receptors initiates constriction of vascular smooth muscle cells.52
Vascular tone is also regulated by hypoxia-independent mechanisms, including the renin-angiotensin-aldosterone system. ACE2 cleaves angiotensin II (AngII) to angiotensin 1–7 (Ang1-7) and downregulation of the ACE2 receptor, following internalisation with SARS-CoV-2, would suppress Ang1-7-mediated vasodilation (figure 2).56 The subsequent accumulation of AngII, and binding to angiotensin II receptor type 1 (AT1), could augment pulmonary vasoconstriction and promote induction of TF and PAI-1 expression on platelets and the endothelium.36 56 57 Increased AngII occurs in COVID-19 and has been associated with viral load and lung injury.58 Imbalance of ACE2/AngII may in part explain the association of pre-existing vascular diseases (eg, systemic hypertension, diabetes) with susceptibility to severe COVID-19 as these diseases have altered baseline levels of ACE2.58
The immune system and thrombosis
Haemostasis and the immune system are intricately related, with the two systems complementing each other to provide host defence and limit the dissemination of invading pathogens. Physiological immunothrombosis can become dysregulated resulting in excessive formation of immunologically mediated thrombi that predominantly affect the microvasculature.15 Immunothrombosis has been proposed as an important pathological mechanism in patients with COVID-19, whereby innate immune cell activation, excessive coagulation and endothelial dysfunction contribute to the observed prothrombotic state.59 Interaction between the haemostatic and innate immune systems, particularly monocytes, macrophages and neutrophils, is the cardinal feature of immunothrombosis (figure 3). Activation of innate immunity can be induced by the coagulation system; thrombin and factor Xa can activate innate immune cells through their protease-activated receptors. Similarly, fibrinogen and fibrin have been shown to initiate the activation of neutrophils.15

{kind=link}
{kind=link}
{kind=link}
Immunothrombosis in COVID-19. Initial binding of SARS-CoV-2 to type II pneumocytes within the alveoli results in mass innate immune cell infiltration (including monocytes, macrophages and neutrophils). Subsequent cytokine release, from these immune cells, contributes to a hypercoagulable state through various proposed mechanisms. (A) Proinflammatory cytokine release can induce the release of platelets and their activation and aggregation. Interleukin (IL)-6, in particular, has been shown to promote the production of platelets with notably more thrombogenic capacity than those produced under a non-inflammatory environment. Cathepsin G, a serine protease produced by neutrophils, also activates platelets. Tumour necrosis factor (TNF)-ɑ and IL-6 upregulate tissue factor (TF) expression by a number of different cell types, namely monocytes, macrophages and endothelial cells. Furthermore, TNF-ɑ triggers a rise in plasminogen-activator inhibitor-1 (PAI-1), which in turn inhibits tissue plasminogen activator (tPA). A consequent reduction in the activity of plasmin reduces fibrinolysis. (B) Complement activation is an important inducer of coagulation. Membrane attack complexes (MACs), also referred to as terminal complement complexes C5b-9, are the end point of a complex cascade of sequential cleavage and activation of complement proteins. MACs are constructed from a number of complement protein subunits and serve as a transmembrane channel, initiating cell lysis of the target cell of which they are embedded. Cell lysis and death of host cells contributes to coagulopathy by initiating microthrombi and von Willebrand factor (VWF) formation, as well as increasing prothrombin activity. Another mechanism, by which complement activation contributes to coagulation, is via binding of C3b to CR1 receptor, present on the membrane of platelets. Binding of C3b triggers the release of short-chain polyphosphate (polyP) from platelets, which induces the expression of TF. Complement component C5a may also contribute to the recruitment of neutrophils. (C) The generation of neutrophil extracellular traps (NETs), as a defence mechanism by neutrophils, also promotes coagulation. Histones, a major component of NETs, attract and bind platelets resulting in their aggregation. Activation of platelets, as a result of their binding to histones, induces TF expression. Neutrophil elastase (NE) cleaves tissue factor pathway inhibitor (TFPI), thereby permitting unprohibited action of TF.
In COVID-19, vascular injury is induced by endocytosis of SARS-CoV-2 by host cells, causing them to undergo pyroptosis.34 Pyroptosis is an extremely inflammatory form of programmed cell death that terminates in cell lysis, causing the release of various DAMPs including ATP, nucleic acids and inflammasomes. Pyroptosis also releases non-encapsulated viral RNA and proteins that can infect surrounding host cells and further amplify the inflammatory milieu. DAMPs bind pattern recognition receptors (PRRs), present on the surface of local epithelial cells, endothelial cells and monocytes.34 Ligation of viral single-stranded RNA and double-stranded RNA (which serve as pathogen-associated molecular patterns (PAMPs)) with PRRs and toll-like receptors (TLRs), on the surface of macrophages, triggers their activation and further exacerbates the proinflammatory response. Recognition of PAMPs through the TLR and CD14 receptor of monocytes promotes the transcription and expression of TF.15 60 The cumulative response of the immune system to SARS-CoV-2, both through inflammation and immune cell expression of prothrombotic proteins, is likely to be a major contributor to hypercoagulability in COVID-19.
Cytokines and chemokines
Severe COVID-19 is characterised by the increased activation of the innate immune system and increased inflammation.34 This is associated with an amplified and uncontrolled release of cytokines, a phenomenon that has been termed cytokine storm.61 Cytokines and chemokines are proteins secreted by a host of immune cells, and serve as an important innate defence mechanism. They recruit adaptive immune cells, and regulate a wide range of processes in the immune system.62 In COVID-19, numerous cytokines and chemokines are increased; IL-6, interferon (IFN)-γ and IL-2 are among the most commonly reported elevated cytokines.61 63 IL-6 increases platelet production and activity, increases the expression of TF on endothelial cells and monocytes and can also give rise to endothelial dysfunction.61 63 IFN-γ similarly increases platelet production and impairs the vascular endothelium, giving rise to prothrombotic effects.63 IL-2 can decrease fibrinolysis by upregulating PAI-1.63 IL-8 is also elevated in COVID-19, and can attract neutrophils to the site of infection, which predisposes to the formation of neutrophil extracellular traps (NETs).61 While cytokines have prothrombotic effects, the degree that this applies to COVID-19 immunothrombosis requires further investigation and reverse causation (immunothrombosis may increase cytokines) or co-association are alternative explanations.
Complement
Complement activation is observed in COVID-19, with the deposition of the terminal complement complex C5b-9 and MASP2 protein in lung lesions.64 Complements are proteins that enhance the function of phagocytic cells and facilitate antibody opsonisation, serving as an important host defence mechanism of the innate immune system. They are produced as dormant factors by the liver, and in COVID-19 they are activated by the alternative and lectin pathways.64 The complement system involves a cascade of processes, culminating in the formation of the terminal C5b-9 membrane attack complex (MAC), which is observed in COVID-19.64 The insertion of a MAC into the cell membrane of infected cells or directly onto pathogens creates a transmembrane channel, triggering cell lysis and death.65 MACs can also activate platelets, induce endothelial secretion of VWF and cause endothelial damage when inserted into endothelial cells.65 When these normal defences against pathogens are hyperactivated, they result in excess endothelial damage that can serve as foci for thrombosis. The individual complement components are prothrombotic, for example, C5a can upregulate the activity of TF and PAI-1 and can also activate neutrophils, resulting in increased IL-6 and IL-8 production, while also promoting the formation of NETs.65 The serine protease MASP2 is increased in COVID-19 and may promote clot formation by activating C2 and C4, which increase the activity of thrombin, fibrinogen and factor XIII.64 66 Complement activation is likely to augment the COVID-19 prothrombotic phenotype and future research should clarify the specific components of the complement system involved and the effect of modulation.
Neutrophil extracellular traps
Neutrophils are important contributors to the formation of thromboses and rapidly migrate to the site of endothelial damage alongside platelets.67 An important defence mechanism, known as NETosis, is deployed by activated neutrophils to clear pathogens and could be relevant to thrombosis in COVID-19 (figure 3).68 NETosis involves the extracellular release of NETs, which are composed of chromatin and microbicidal proteins.69 NETs have been implicated in the pathobiology of thrombosis in VTE, as well as ARDS and sepsis, with serum levels of NETs correlating with mortality.70 71
NET-driven thrombosis is largely platelet-dependent; neutrophils recognise and bind P-selectin, expressed by activated platelets, through their PSGL-1 receptor which triggers NETosis.67 NETs can also interact with VWF, released by endothelial cells and platelets, which leads to platelet adhesion and fibrin formation.72 73 Histone proteins in the DNA fragments of NETs, serve as potent DAMPs which can further attract platelets and thereby initiate a positive feedback loop.67 Release of neutrophil elastase, a serine protease, during NETosis has previously been shown to inhibit anticoagulation by degrading TFPI and thrombomodulin.74 Degradation of these endogenous anticoagulants permits the unprohibited action of TF.75 Serine proteases also degrade alveolar surfactant cells which are important in the clearance of inflammatory cells.71
The detrimental effects of NETs have previously been described in sepsis and ARDS, whereby NETs have been shown to induce damage to host tissue at the site of injury, thus exacerbating local inflammation and propagating microvascular thrombosis. Case reports in severe COVID-19 have described evidence of NETs, with sera derived from hospitalised patients containing markers of NETs, including elevated levels of citrullinated histone H3 and myeloperoxidase-DNA.68 A study using autopsy-derived tissue from patients with COVID-19 reported neutrophil activation and the presence of NET aggregates within the microvasculature, resulting in vascular occlusion and consequent organ damage.76 NET formation may be augmented by the described proinflammatory and procoagulant factors in COVID-19, contributing to a thrombotic phenotype.
COVID-19 and VTE genetic associations
VTE is a polygenic disease associated with common and rare genetic variants including heritable thrombophilias (eg, factor V Leiden, prothrombin mutations and antithrombin, protein C/S deficiencies). A genome-wide association study (GWAS) of patients with severe COVID-19 identified genetic associations in the ABO gene and in a chromosome 3 locus (3p21.31) spanning several genes (SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6 and XCR1).77 When ABO blood groups were inferred in this GWAS, the A group was enriched in the patients with severe COVID-19 while the O group was under-represented. ABO is a pleiotropic locus that is associated with thrombotic diseases including VTE.78 The differential thrombotic risk of ABO blood groups has traditionally been attributed to VWF levels, which are 25% lower in O group individual.79 ABO is also associated with IL-6 levels which, together with VWF, are increased in COVID-19.80 Alternatively, anti-A antibodies in blood group O and B individuals may inhibit the SARS-CoV-2 virus and ACE2 receptor interaction.81 82 Multiple plasma protein levels are associated with ABO, relating to immunology, endothelial cell function and coagulation.83 Alternative functional consequences of ABO genetic variation include influencing DC-SIGN levels, a membrane receptor expressed by dendritic cells and a proposed binding site for SARS-CoV-2.84 85
The candidate gene SLC6A20 in the 3p21.31 GWAS locus encodes the SIT1 transporter protein that functionally interacts with the ACE2 receptor, and the 3p21.31 locus is putatively associated with levels of the chemokine CXCL16; however, the functional consequences of the 3p21.31 genetic association require further investigation.77 84 85 Genetic variation in ACE2 and TMPRSS2 may influence COVID-19 susceptibility but this requires validation in a COVID-19-specific population.86 Genetic variation in human leucocyte antigens could affect immune response by varying the affinity for SARS-CoV-2 binding.87 Furthermore, rare variants in genes related to type I IFN immunity are enriched in severe COVID-19.88Ongoing research from global consortia aims to clarify the role of genomic variation in COVID-19 susceptibility, severity and clinical outcomes.89
Additional thrombotic mechanisms
Additional mechanisms have been putatively associated with thrombosis in COVID-19. Increased levels of ferritin in COVID-19 are likely to reflect cellular damage and could contribute to inflammation.90 91 High levels of ferritin may have detrimental effects on mitochondria, leading to the release of reactive oxygen species, which cause cell death.91 Mitochondrial dysfunction in platelets may contribute to inflammation and a prothrombotic state.91
Elevated antiphospholipid antibody (APA) titres have been described in COVID-19, although their significance is unclear.92 APAs can interact with the endothelium, leucocytes and platelets, triggering the release of prothrombotic factors and can also interact with the complement system.93 APAs can be raised in acute infection, and a diagnosis of antiphospholipid syndrome requires APAs to be measured on two separate occasions 12 weeks apart, which needs confirmation before being implicated in COVID-19 pathophysiology.92 93
Obesity is a long-term and subacute inflammatory condition that is a risk factor for COVID-19 and VTE.36 94 95 Hypertrophy of adipocytes and the associated dysfunction in adipose metabolism causes the release of IL-6, PAI-1 and TF, which activate the coagulation system.95 Platelet aggregation is also promoted with the decreased release of adiponectin and increased release of leptin.95 Insulin resistance, associated with obesity, also reduces the modulatory effect that insulin appears to have on platelet activity.95 The inflammatory state in obesity may account for its association with COVID-19, and result in an increased risk of VTE.
Discussion
The mechanisms contributing to increased thrombosis in COVID-19 involve extensive cross-talk between haemostasis and the immune system. Treatments that target these pathways may mitigate the adverse macrovascular and microvascular effects of COVID-19 and include anticoagulants, antiplatelets, fibrinolytics and immune modulators, with numerous studies ongoing.96 Guidelines recommend prophylactic anticoagulation in hospitalised patients with COVID-19 and treatment dose anticoagulation in established VTE, with evidence indicating better clinical outcomes for anticoagulated patients.20 97 Ongoing studies are assessing different anticoagulation strategies for COVID-19. Anticoagulation may reduce propagation or additional formation of thrombus, but alternative strategies may be required to prevent or target dysregulated immunothrombosis. Additionally, cellular heparan sulfate is a proposed co-receptor for SARS-CoV-2 binding to ACE2, and therefore exogenous heparin may have effects on viral adhesion.98 Dexamethasone has a range of anti-inflammatory and immunosuppressive effects including attenuating the function of immune cells, particularly T cells by suppressing their activation and proliferation. Dexamethasone improves clinical outcomes in hospitalised patients with COVID-19, but its role in controlling COVID-19 immunothrombosis is unclear.99 100 Fibrinolytics (eg, recombinant tPA) have been trialled in a case-series of COVID-19 ARDS, and tPA may have additional anti-inflammatory effects that could be beneficial for COVID-19 immunothrombosis.43 Anticytokine treatments, such as tocilizumab (directed against the IL-6 receptor), and anticomplement agents, such as eculizumab (directed against C5), are also being investigated in patients with COVID-19.61 Attenuating proinflammatory pathways is likely to have downstream effects on immunothrombosis. However, care is required to strike the correct balance between appropriately targeting aberrant and dysregulated immunothrombosis, while not impairing its important physiological host defence function.
While research into COVID-19-associated VTE and immunothrombosis has been proliferating, there remain a number of knowledge gaps. Some putative pathobiological mechanisms have been inferred from other disease processes including alternative betacoronaviruses, viral pneumonias and ARDS. Dissecting and delineating the specific effects that SARS-CoV-2 has on thrombosis remains an active area of research and is crucial for guiding interventions. Areas for future COVID-19 research include (i) whether small pulmonary thromboses represent VTE, immunothrombosis or a combination; (ii) can diagnostic strategies (eg, radiological, biochemical) accurately diagnose and differentiate between VTE and immunothrombosis; (iii) can the risk of immunothrombosis be accurately predicted; (iv) can immunothrombosis be prevented with prophylactic anticoagulation, or treated with anticoagulation; (v) developing novel immunothrombosis targeted interventions and defining how other COVID-19 treatments (eg, dexamethasone) affect immunothrombosis/VTE in COVID-19.
Most COVID-19 studies have been cross-sectional in patients with more severe disease. To fully understand the immuno-haemostatic cross-talk leading to immunothrombosis, longitudinal measurements in different cohorts would be required, which would guide the optimal timing and cohorts where intervention would be beneficial. An increasing understanding of the complex pathobiological interplay between the immune system and haemostasis in COVID-19 will help in developing new treatments and mitigate off-target effects of modulation.
References
Footnotes
Contributors JL undertook the literature search, co-wrote the first draft and prepared the final draft. DAS co-wrote the first draft and prepared the final draft and figures. MN conceived the idea for the manuscript, co-wrote the first draft and prepared the final draft.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.