Article Text

Abstract
Rationale The most common antibiotic used to treat people with cystic fibrosis (PWCF) is inhaled tobramycin, administered as maintenance therapy for chronic Pseudomonas aeruginosa lung infections. While the effects of inhaled tobramycin on P. aeruginosa abundance and lung function diminish with continued therapy, this maintenance treatment is known to improve long-term outcomes, underscoring how little is known about why antibiotics work in CF infections, what their effects are on complex CF sputum microbiomes and how to improve these treatments.
Objectives To rigorously define the effect of maintenance tobramycin on CF sputum microbiome characteristics.
Methods and measurements We collected sputum from 30 PWCF at standardised times before, during and after a single month-long course of maintenance inhaled tobramycin. We used traditional culture, quantitative PCR and metagenomic sequencing to define the dynamic effects of this treatment on sputum microbiomes, including abundance changes in both clinically targeted and untargeted bacteria, as well as functional gene categories.
Main results CF sputum microbiota changed most markedly by 1 week of antibiotic therapy and plateaued thereafter, and this shift was largely driven by changes in non-dominant taxa. The genetically conferred functional capacities (ie, metagenomes) of subjects’ sputum communities changed little with antibiotic perturbation, despite taxonomic shifts, suggesting functional redundancy within the CF sputum microbiome.
Conclusions Maintenance treatment with inhaled tobramycin, an antibiotic with demonstrated long-term mortality benefit, primarily impacted clinically untargeted bacteria in CF sputum, highlighting the importance of monitoring the non-canonical effects of antibiotics and other treatments to accurately define and improve their clinical impact.
- cystic fibrosis
- respiratory infection
- bacterial infection
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The accession number for all metagenomic sequencing and 16S amplicon sequencing data reported in this paper is NCBI Bioproject: PRJNA530252.
Statistics from Altmetric.com
Key messages
What is the key question?
What are the effects of chronic inhaled tobramycin therapy on the cystic fibrosis (CF) sputum microbiota and metagenome?
What is the bottom line?
Using next generation metagenomic sequencing, we found that tobramycin overwhelmingly affected sputum abundances of non-dominant, prevalent taxa after 1 week of therapy and that these changes often rebounded after antibiotic pressure is removed. Community functional capacity changed relatively little, however, suggesting functional redundancy despite taxonomic plasticity.
Why read on?
Tobramycin maintenance therapy has been demonstrated to reduce exacerbation frequency and improve mortality in people with CF; however, the underlying microbiological effects and effectors of this therapy still remain poorly defined.
Introduction
People with cystic fibrosis (PWCF) have chronic respiratory infections that require frequent antibiotic treatments. While these infections contribute considerably to reduced quality and length of life,1 2 neither the microbial determinants of CF respiratory disease severity nor of response to antibiotic treatment are well understood. While standard culture techniques usually yield a small number of classic pathogens such as Staphylococcus aureus and Pseudomonas aeruginosa in CF sputum, a commonly collected respiratory specimen that variably samples the entire respiratory tract,3 4 culture-free sequencing methods frequently identify diverse, uncultured taxa. These newer techniques also suggest CF sputum microbiota are relatively stable, with only transient changes during exacerbations and antibiotic treatments.5 6 While one might predict that antibiotic-induced reductions in sputum abundances of ‘pathogenic’ bacteria (such as P. aeruginosa) are responsible for clinical improvement, previous studies have not supported this hypothesis,5 6 usually identifying minimal and/or transient abundance changes during treatment or exacerbations that do not consistently correlate with changes in either symptoms, lung function or in vitro clinical susceptibility testing.7 8 Conversely, the presence in CF sputum of diverse microbiota has been associated with better clinical status,9 10 but the mechanisms of these associations and their roles in therapeutic responses are unclear. For example, higher sputum microbial diversity may signify lower historical antibiotic burden due to milder disease; alternatively, higher diversity could indicate low absolute abundances of pathogens that only lead to worse disease when their abundances rise. Furthermore, while most CF microbiome research has focused on the microbial taxa (‘microbiota’) in sputum, the full complements of sputum microbial genes (‘metagenomes’) have not been defined. Metagenomic analysis provides an opportunity to study several poorly understood features of CF lung disease, including the mechanisms underlying the resilience of CF respiratory infections, during potent antibiotic treatments.11–14
Inhaled tobramycin is the antibiotic prescribed most often for CF lung disease15 and has been demonstrated to decrease sputum P. aeruginosa abundances and improve long-term respiratory outcomes for PWCF on average when used in 28 day, alternate month cycles.11 16 17 However, studies have shown the short-term effects of tobramycin observed in treatment-naive patients—reduction in P. aeruginosa counts and improved respiratory function—are attenuated with successive drug cycles11 and vary between patients regardless of the in vitro susceptibilities of infecting P. aeruginosa to tobramycin.11–14 Tobramycin is an aminoglycoside, a class of bactericidal antibiotics that act at bacterial ribosomes to inhibit protein synthesis and promote the production of toxic defective proteins.18 Aminoglycoside antibiotics have been shown to have in vitro activity against other common CF pathogens, including gram-negative organisms19 and S. aureus,20 in addition to P. aeruginosa. Together, this spectrum of aminoglycoside activity and the known discrepancies between microbiological effects and clinical outcomes suggest that factors beyond Pseudomonas, perhaps involving other taxa or specific microbial genes, could contribute to clinical and microbiological responses to this therapy.
We hypothesised that changes in CF sputum microbiomes (both microbiota and metagenomes) with inhaled tobramycin therapy would involve taxa beyond the intended target, P. aeruginosa, and that defining these microbiome dynamics would identify candidate mechanisms by which chronic infections in CF persist despite antibiotic treatment. Many previous studies have been limited by unstandardised study designs and heterogeneous subject characteristics, making inferences from microbial analysis and clinical outcomes difficult.21–24 Thus, to test our hypotheses, we analysed sputa collected from PWCF surrounding a standardised, routine maintenance course of inhaled tobramycin using culture, quantitative PCR (qPCR) and metagenomic sequencing, and comparing microbiological changes during treatment with changes in pulmonary function measures and subjective symptom scores. Our study focused on the effects of this treatment during routine maintenance medication; the work by Heirali et al complements this study by analysing the effects on a treatment-naive study population. Our goal was to specifically investigate the taxonomic and functional21–24 changes with a single course of maintenance inhaled tobramycin and to identify the determinants of sputum microbial persistence during antibiotic treatment in CF in order to direct the development of more focused and effective therapies for these and many other recalcitrant infections.
Results
Lung function and symptom scores changed minimally with inhaled tobramycin
We collected spontaneously expectorated sputum samples from 30 PWCF before (‘baseline’), weekly during (‘weeks 1–4’) and 1 month after (‘follow-up’) a standard 28-day course of tobramycin inhaled powder (TIP), comprising 157 samples. Subjects had a mean age of 35 years (range 9–75) and most (24/30) subjects reported routine use of these treatments (further details in online supplementary table S1). Subjects received no antibiotics other than maintenance azithromycin for ≥1 month prior to or during the study, and subjects were withdrawn if they required other antibiotics during the study period. Lung function measurements (FEV1 % predicted, ppFEV1) and symptom scores (Chronic Respiratory Infection Symptom Score, CRISS) were collected (online supplementary figure S1). Consistent with previous work,11 most participants’ lung function measurements demonstrated little change with maintenance inhaled tobramycin treatment. We found a significant negative association between ppFEV1 and CRISS score, as well as between changes in these two metrics, with treatment (online supplementary figure S1C,D), indicating that improvement in lung function was associated with improvement in symptoms.
Supplemental material
Changes in clinical metrics did not correlate with changes in sputum microbiota with inhaled tobramycin
Of the 30 study participants, 26 provided both baseline and week 4 sputum samples and performed spirometry. We compared changes in ppFEV1 and CRISS scores with both baseline sputum microbiological measures and changes in those measures during therapy, including absolute cultured abundances of S. aureus and P. aeruginosa, qPCR-defined total bacterial load (TBL) and qPCR-defined absolute abundances of specific bacteria, sequencing-defined relative abundances of individual taxa, diversity measures and normalised abundances of functional gene categories. As expected for this clinically stable population during maintenance therapy, we identified no significant associations between microbiological and clinical measures. We repeated this analysis post hoc using percent change in ppFEV1 as a measure of clinical outcome (in contrast to absolute change from baseline) and found similar results (data not shown). We therefore focused on comprehensively defining how sputum microbiology changes with a cycle of maintenance inhaled tobramycin.
Primary sputum microbiological changes occurred at 1 week of inhaled tobramycin, with marked inter-individual variability
Culturable sputum abundances of lactose-nonfermenting, gram-negative bacteria (predominantly P. aeruginosa) and of S. aureus decreased on an average of 1.7 and 1.1 logs, respectively, at 1 week of therapy. Viable counts of both plateaued or dropped relatively little after week 1, increasing thereafter during treatment, suggesting adaptation to antibiotic pressure that limited antibacterial efficacy (figure 1A–D). We did not identify any strain switching among P. aeruginosa or S. aureus using metagenomic sequencing (online supplementary figure S4), indicating that drops in sputum viable counts with tobramycin could not be explained by selecting for or against specific strains. To investigate whether change in viable counts is a better measure of tobramycin therapeutic success than spirometry or symptom score, we compared drop in sputum viable count after 1 week in both P. aeruginosa and S. aureus to relative abundances of individual taxa, diversity measures and normalised abundances of functional gene categories as described above. We identified no significant associations between these molecular microbiological measures and change in viable counts of canonical CF pathogens.

Intersubject variability of sputum microbiological responses to a cycle of maintenance inhaled tobramycin was greater than intrasubject variability. (A and B) Change in culturable colony counts on MacConkey agar (sequencing-based analyses demonstrated these to be predominantly Pseudomonas aeruginosa with minor contributions of Serratia marcescens, Stenotrophomonas maltophila, Achromobacter xylosoxidans and an unidentified yeast taxon) from baseline (A) and absolute viable counts (B) by week on therapy. (C and D) Similarly show change in viable counts on mannitol salt agar (Staphylococcus aureus) from baseline (C) and absolute viable counts by week on therapy (D). (E and F) Change in TBL from baseline (E) and absolute TBL, (F) by week on therapy. Black lines represent individual subjects and coloured lines indicate medians. Baseline samples were collected prior to starting therapy, weeks 1–4 represent weekly samples collected during therapy and follow-up samples were collected 1 month after cessation of therapy. P values were determined using a Wilcoxon signed rank sum test comparing baseline and week 1 values. All values are presented after log transformation. TBL, total bacterial load.
To define total sputum microbiota dynamics, focusing on viable cells, we extracted DNA from all samples using a method that depletes human and extracellular bacterial DNA25 prior to sequencing. In contrast to culture abundance, the mean absolute bacterial load, defined by broad-range qPCR, changed ≤0.5 logs with therapy (figure 1E). The different magnitudes of changes in TBL (figure 1E,F) and in classic pathogen culture counts (figure 1A–D) could in part be attributable to increased abundances of viable but unculturable cells, a state that can be induced by antibiotic exposure26 27 and that could offset drops in DNA-based absolute abundances of canonical CF pathogens. Alternatively, DNA from dead but intact cells could have impacted PCR-based findings. To further investigate these questions at the species level, we used quadruplex qPCR to measure absolute abundances of four taxa exhibiting changes during therapy (online supplementary figure S2B): P. aeruginosa, S. aureus, Prevotella spp and Streptococcus spp (online supplementary figure S2). While P. aeruginosa and S. aureus dynamics defined by qPCR were qualitatively similar to those observed from culture, the magnitudes of their qPCR-defined changes were smaller than those defined by culture. Notably, qPCR-defined Prevotella and Streptococcus abundances changed little, suggesting that absolute abundance increases in these taxa did not account for minimal TBL changes, and suggesting important contributions of viable, non-culturable cells and/or extracellular DNA to CF sputum microbiota, as suggested previously.25 28 29
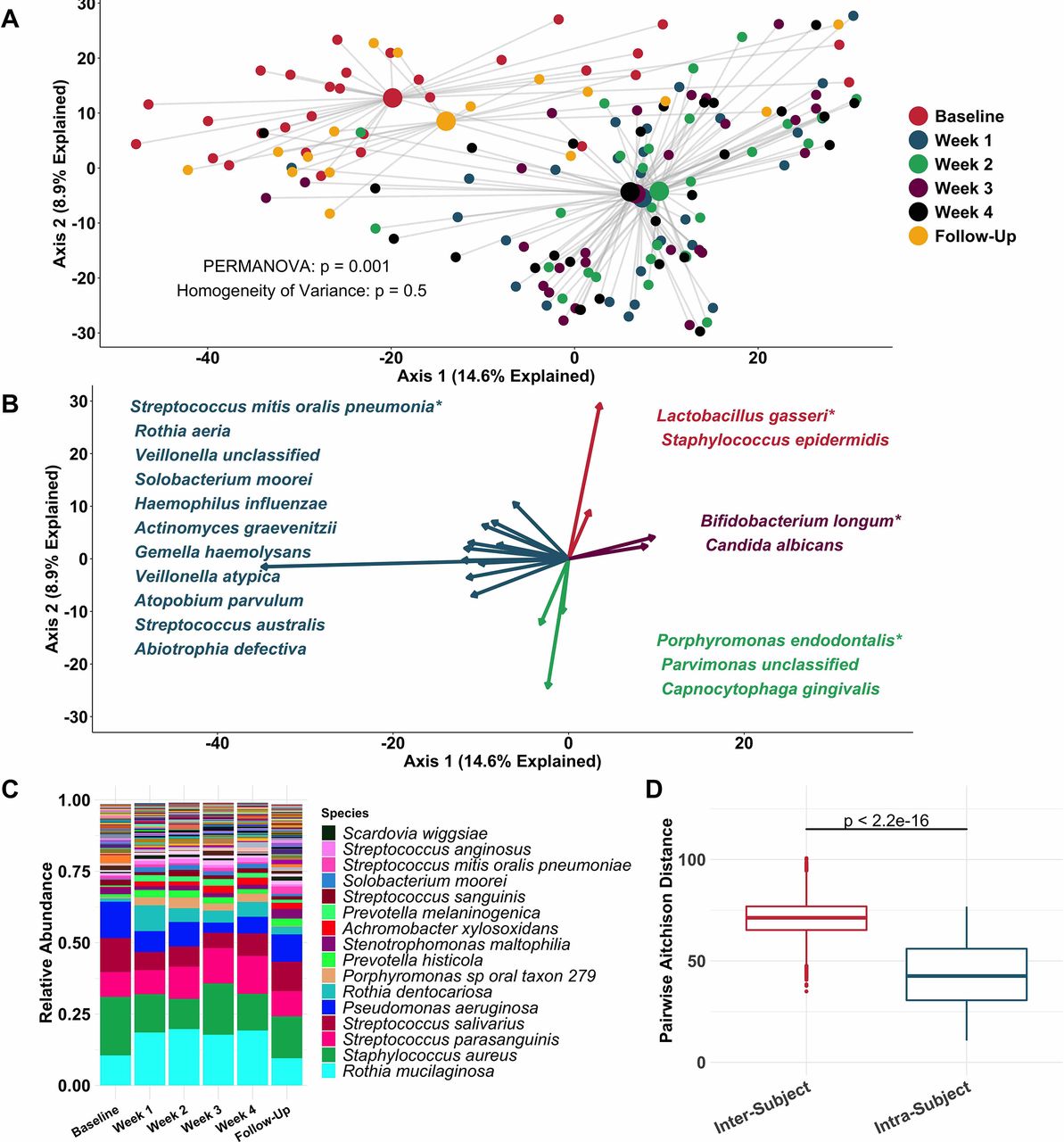
Principal coordinate analysis (figure 2A) identified clear distinctions among sputum bacterial microbiota between samples collected while off (baseline and follow-up samples), compared with on (weeks 1–4) tobramycin. PERMANOVA indicated sputum microbiota differed significantly by week on therapy (figure 2A), while Homogeneity of Variance analysis demonstrated that these differences (distances between centroids, figure 2A) were not explained by differences in microbiota variances by week (distances between individual datapoints and their respective centroids). Therefore, the majority of sputum taxonomic changes with maintenance inhaled tobramycin occurred at 1 week of therapy. These treatment-emergent changes in sputum microbiota were most marked among taxa other than classic CF pathogens such as P. aeruginosa and S. aureus (figure 2B,C).
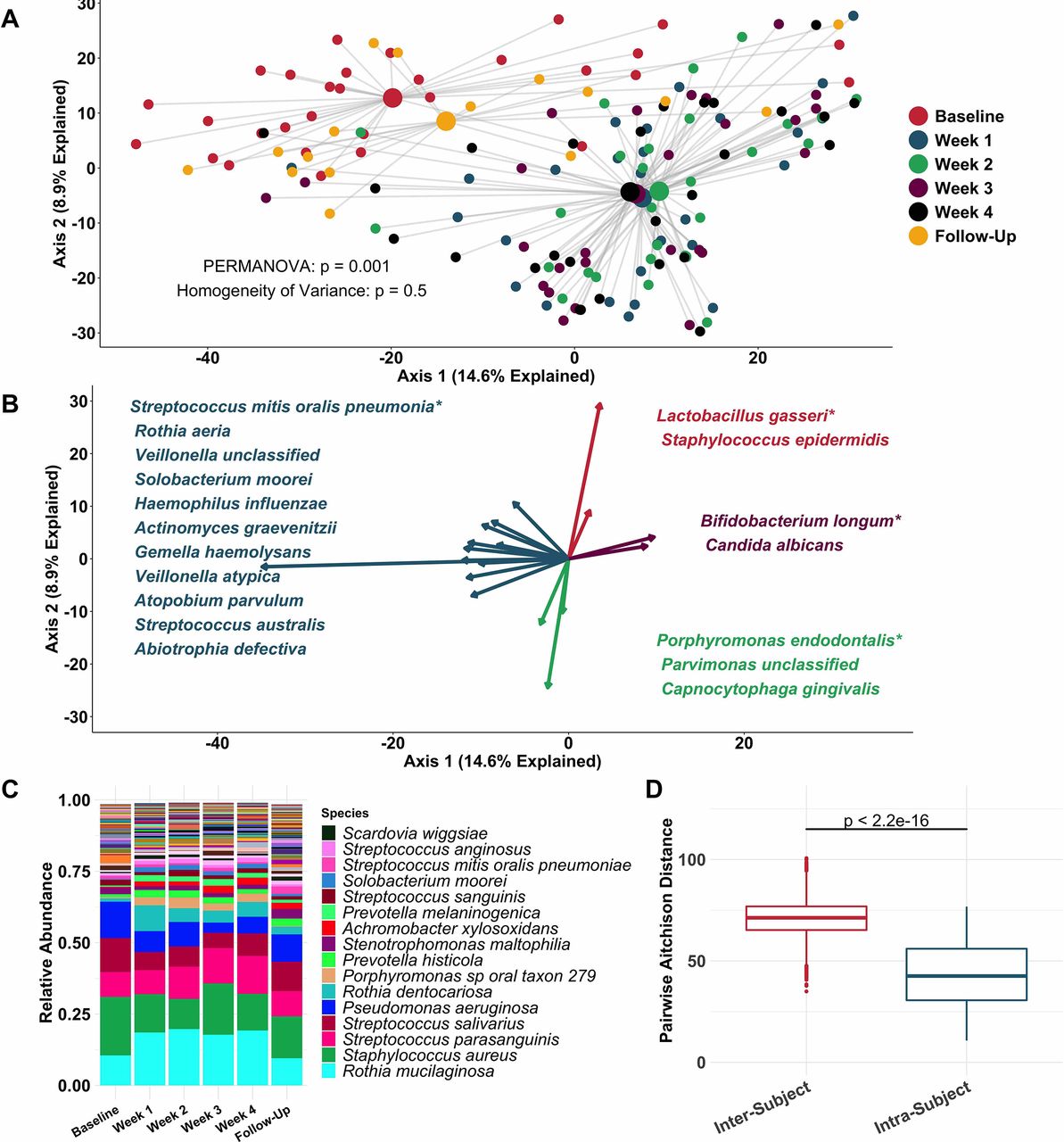
Sputum microbiota shifts after 1 week of inhaled tobramycin. Taxonomic profiles of all samples were calculated via metagenomic shotgun sequencing followed by metagenomic phylogenetic analysis. (A) Principal components analysis using the Aitchison dissimilarity metric—pairwise Euclidean distance between samples after a centred log-transformation of relative abundance data, which is optimised for sparse, compositional data such as microbiota61 62—of all samples (small dots) coloured and grouped by week on therapy. Large dots represent the centroid of each group, with lines connecting individual sample dots to their respective centroids. PERMANOVA tested for difference between centroids, Homogeneity of Variance assessed whether dispersion in data within each timepoint (distance of each datapoint from the respective centroid) differed among groups. (B) Biplot demonstrating the 18 taxa most responsible for the taxonomic difference between samples in (A). Length of vectors indicates the extent to which taxa contribute to intersample dissimilarity; starred taxa are those with longest vectors in each colour grouping. (C) Average taxonomic profiles of all samples by week on therapy at the species level. Only the top 14 most abundant species names are shown for ease of display. (D) Comparison of intrasubject versus intersubject microbiota dissimilarity at the species level by Aitchison dissimilarity, which takes into account both abundances and presence of individual taxa. Baseline samples were collected prior to starting therapy, weeks 1–4 represent weekly samples collected while individuals were on therapy and follow-up samples were collected 1 month after cessation of therapy. Wilcoxon signed-rank test was used to assess difference between groups. Taxonomic profiles of all samples individually are presented in online supplementary figures S4 and S5. Boxes represent interquartile region and middle represents the median. Streptococcus mitis, Streptococcus oralis and Streptococcus pneumoniae cannot be reliably differentiated by MetaPhlAn2 and are grouped in this analysis.
Inspection of individual, sequencing-based taxonomic data at the genus and species levels (online supplementary figures S5 and S6) demonstrated marked intersubject variability in these treatment-emergent changes. To quantify this observation, we defined the diversity of sputum microbiota both within and between subjects, demonstrating much higher diversity between than within subjects (figure 2D): Sputum taxonomic shifts with antibiotic perturbation were of a smaller magnitude than differences between any two samples from different subjects. Because metagenomic sequencing on a large scale has rarely been used previously to define CF sputum microbiota, we also sequenced a portion of samples with the more commonly used 16S amplicon sequencing, yielding nearly identical results, although at the genus level compared with the species-level resolution of metagenomic analysis (online supplementary figure S7). We also calculated change in Shannon and Simpson diversity indices, evenness and richness for all samples by week on therapy (online supplementary figure S3). Despite substantial intersubject variability in these measures, we observed a statistically significant decrease in average species richness after 1 week of therapy, returning to baseline levels on follow-up. We identified no association between microbial read depth and Shannon or Simpson index, and a paradoxical negative association between species richness and microbial read depth, indicating that sequencing depth did not impact these results (online supplementary figure S8).
Sputum taxonomic shifts are driven by non-dominant, facultative and obligate anaerobes
To more rigorously identify which taxa changed most in sputum abundance with maintenance tobramycin treatment, we compared the sequencing-defined microbiota at baseline with those at week 1, when most changes occurred (figure 2A). Although P. aeruginosa was the intended target of inhaled tobramycin therapy,11 we found the most significant sputum relative and calculated absolute abundance changes occurred among non-dominant, low abundance taxa (figure 3B,C). In fact, no classic CF pathogen abundance changed significantly with tobramycin by molecular analysis (figure 3B,C, online supplementary figure S9), although the calculated abundance changes of both P. aeruginosa and S. aureus at week 1 were similar to those we identified using traditional culture (figure 1B,D vs figure 3C). To determine how specific taxa related to clinical response, we repeated the above analysis after partitioning subjects into dichotomous ‘responder’ versus ‘non-responder’ categories (wherein ‘responders’ were subjects in the upper tertile of ppFEV1 change; >0 percentage point increase). We identified no significant differences between groups in either baseline sputum microbiota (figure 4A) or their change after week 1 (figure 4B); parallel analyses identified no associations with symptomatic responses (not shown). Interestingly, we did observe trends towards correlations between responder status and higher baseline sputum abundances of P. aeruginosa, despite minimal change in this species with therapy (figure 4C,D).

Non-dominant taxa contribute substantially to taxonomic shift with therapy. Taxonomic profiles of all samples defined by metagenomic sequencing and phylogenetic analysis. (A) Principal component analysis using the Aitchison dissimilarity metric of all baseline and week 1 samples (small dots) coloured and grouped by antibiotic treatment status. Large dots represent the centroid of each treatment category, with lines connecting individual sample dots to their respective centroids. (B) Log-relative abundances and (C) calculated log-absolute abundances of the 15 taxa that contributed most to the differences between baseline and week 1 samples identified in (A) as well as Pseudomonas aeruginosa and Staphylococcus aureus for comparison. PERMANOVA is a statistical test for difference between centroids, Homogeneity of Variance is a statistical test which assessed the difference in spread between two groups. Absolute abundances were calculated by multiplying relative abundances via MetaPhlAn2 by total bacterial loads as determined via universal 16S qPCR. Boxes represent interquartile regions and middle lines represent the medians. qPCR, quantitative PCR.
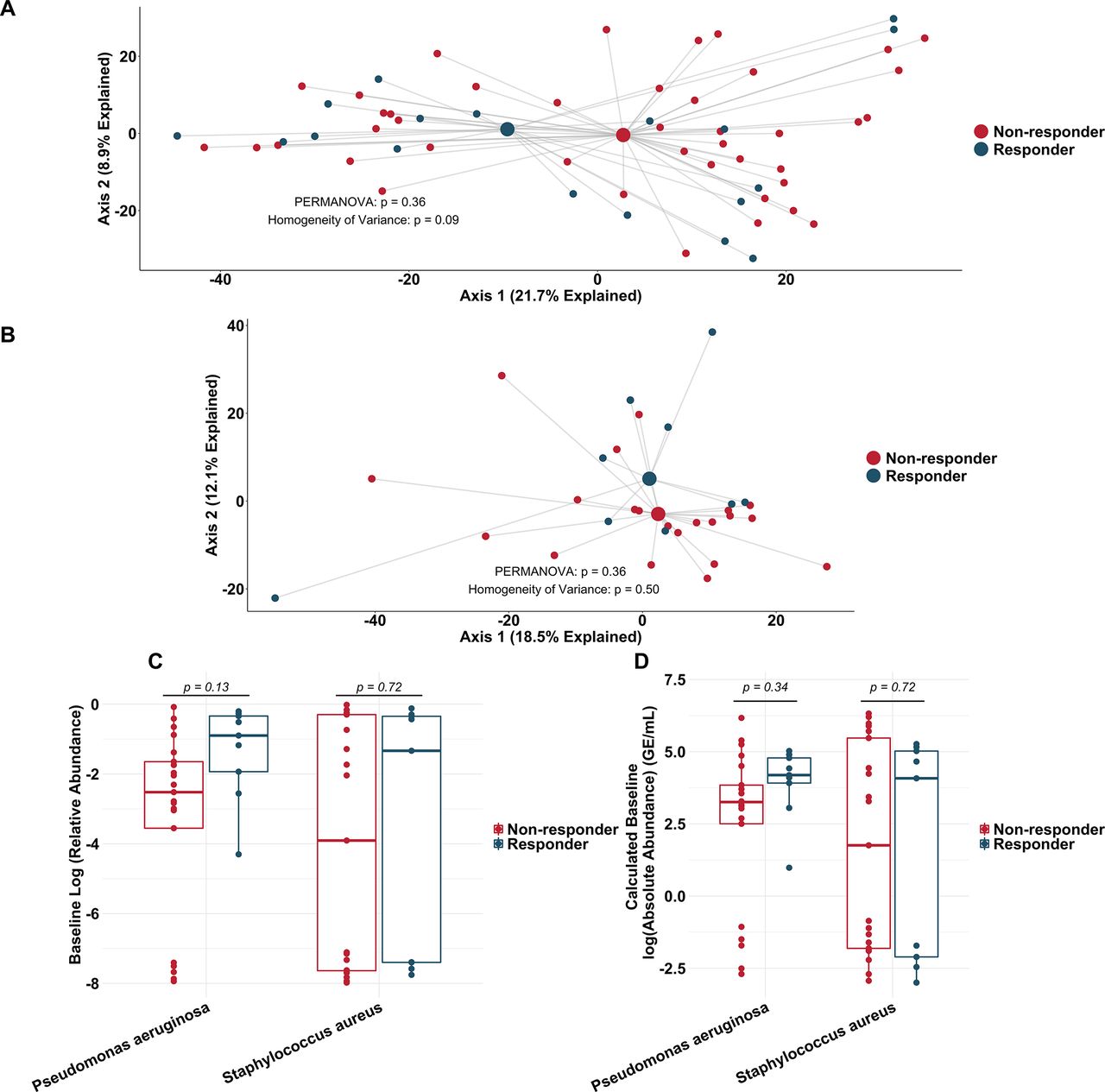
Similarity in sputum microbial communities between ‘responders’ and ‘non-responders’ to tobramycin. (A) Principal component analysis using the Aitchison dissimilarity metric of all baseline samples (small dots) coloured and grouped by response status. Large dots represent the centroid of each group, with lines connecting individual sample dots to their respective centroids. (B) Principal component analysis using the Aitchison dissimilarity metric of changes in relative abundance between baseline and week 1 samples (small dots) coloured and grouped by response status. Large dots represent the centroid of each group, with lines connecting individual sample dots to their respective centroids. PERMANOVA is a statistical test for difference between centroids, Homogeneity of Variance is a statistical test which assessed the difference in spread between two groups. (C–D) Difference in relative abundance (C) and calculated absolute abundance (D) of Pseudomonas aeruginosa and Staphylococcus aureus at baseline between ‘responders’ and ‘non-responders’. Boxes represent interquartile region and middle lines represent the medians.
CF sputum microbial predicted functional capacity is largely maintained with inhaled tobramycin therapy despite changes in taxonomic constituency
We next determined whether antibiotic treatment resulted in changes in sputum metagenomes. We found no significant difference in functional gene content between samples collected at baseline compared with week 1 samples (figure 5A), suggesting that while the taxonomic composition of CF sputum microbiota may shift with antibiotic therapy, functional redundancy dampened metagenomic changes (figure 3A vs figure 5A). However, 13 individual functional gene categories (modules) differed significantly in abundance between samples collected on and off therapy (figure 5B, table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genetically conferred functional capacity changes relatively little with antibiotic therapy. Genetically conferred functional capacity was determined by mapping all shotgun sequencing reads to the KEGG database at the module level. (A) Principal component analysis using the Aitchison dissimilarity metric—Euclidean distance between samples after a centred log-transformation of relative abundance data—of baseline and week 1 samples without removal of dominant taxa (small dots) coloured and grouped by antibiotic status. (B) Log-abundances of all significantly different modules between baseline and week 1 samples without dominant taxa removed. (C) Principal component analysis using the Aitchison dissimilarity metric of all baseline and week 1 samples coloured and grouped by antibiotic treatment status after removal of reads from dominant taxa. (D) Log-abundances of the 20 modules with the largest effect size of those significantly different modules between baseline and week 1 samples after removal of reads from dominant taxa. PERMANOVA is a statistical test for difference between centroids, Homogeneity of Variance is a statistical test which assesses the difference. Modules are listed in table 1. (E) Principal component analysis using the Aitchison dissimilarity metric of all baseline and week 1 samples (small dots) coloured and grouped by antibiotic treatment status using only reads from dominant taxa. Large dots represent the centroid of each group, with lines connecting individual sample dots to their respective centroids. Boxes represent interquartile region and middle represents the median.
Description of functional features shown in figure 5
The contributions of dominant taxa to metagenomic data can mask those of less abundant taxa. To further investigate whether the sputum abundance changes we observed in non-dominant taxa with tobramycin therapy (figure 3B,C) resulted in predicted functional changes among those unconventional microbiota, we computationally removed all reads from dominant taxa, defined here as any taxon with relative abundance twice that of the next most dominant taxon at baseline in any subject,30 and repeated the functional analysis. Significant differences occurred between the predicted functional capacities of non-dominant microbiota in sputum samples collected on and off therapy (figure 5C,D, table 1), primarily among efflux pump and stress response genes, both of which are known to be involved in bacterial responses to antibiotics.31 32 Notably, all modules identified as significantly different after 1 week of therapy with dominant taxa present remained significantly different after removal of dominant taxa, indicating that non-dominant taxa largely drove the observed changes in microbial functional capacity with tobramycin treatment. In support of this interpretation, repeating the analysis in figure 5A using only reads from dominant taxa identified no significant differences between samples collected on and off therapy (figure 5E).
Discussion
Using metagenomic sequencing, qPCR, and standard culture, we defined the CF sputum microbiome response to a cycle of maintenance inhaled tobramycin treatment. While this therapy is primarily intended to target P. aeruginosa, the most significant abundance changes with treatment occurred among non-dominant taxa, none of which are considered classic CF pathogens. Using metagenomic sequencing, a method rarely used in clinical microbiological analyses, we also found that the predicted functional microbial consequences of this treatment involved efflux pumps and response to environmental stresses, again largely among clinically non-targeted and non-dominant taxa, providing clues to the remarkable resilience of CF sputum microbiota to this and other antibiotic treatments. As expected in this study of maintenance treatment, the study population experienced modest changes in both objective (spirometry) and subjective (symptom score) clinical measures during treatment; accordingly, we identified no significant correlations between clinical and microbiological responses in this study. These findings suggest that the primary sputum microbiological effect of tobramycin maintenance therapy is on non-targeted bacteria, offering avenues both to better define the microbial determinants of clinical response to therapy and to devise more effective treatment strategies for a broad range of PWCF and other chronic infections.
The majority of the sputum microbiological change occurred by the first week of therapy. By culture, absolute sputum abundances of traditional CF pathogens (S. aureus and gram-negative organisms, predominantly P. aeruginosa) changed most by week 1 and were followed by either a plateau or gradual return towards baseline levels, suggesting gains made earlier during treatment subsequently diminished. Sputum microbial changes identified by molecular methods were qualitatively similar to culture, but with lower magnitudes; absolute sputum loads of all bacteria and of common taxa (S. aureus, P. aeruginosa, Streptococcus spp and Prevotella spp) changed very little with treatment. Quantitative discrepancies between culture and molecular analyses of CF sputum have been observed before33 and could be attributable to viable, nonculturable bacteria (undermining the accuracy of culture) or DNA from dead cells (particularly problematic for qPCR and sequencing). Our use here of a method that depletes extracellular DNA, including that from host and lysed bacterial cells, mitigates the latter contribution. Further research will be required to quantify and distinguish the contributions of these two effects; however, prior work showing that antibiotics can induce bacteria to enter unculturable states34 suggests this mechanism could contribute to our observations.
As found in studies of CF exacerbations, the sputum microbiota in this population differed more between subjects than within subjects, even during antibiotic treatment. Nevertheless, we identified significant, treatment-emergent changes in sputum microbiomes that were not evident in prior studies, likely due to our study design focusing on a single antibiotic and a defined treatment and specimen collection schedule.5 6 Surprisingly, non-dominant, prevalent taxa contributed most to sputum taxonomic shifts with maintenance tobramycin treatment. Non-dominant taxa therefore represent a conserved feature among CF sputum microbiomes; while the taxa involved differed between individuals, their presence and dynamic responses to maintenance tobramycin varied much less than did those of the dominant taxa. Accordingly, we observed a shift in genetically conferred functional capacity, again primarily at 1 week of therapy and driven largely by non-dominant taxa, but of a much smaller magnitude than that seen in taxonomic constituency, implying functional redundancy and stability within sputum microbiota. This relationship between taxonomic and functional microbiome characteristics has been observed for diverse microbial communities,35 36 suggesting CF sputum microbiota share characteristics with other well-established microbiota.
The non-dominant taxa that shifted most in abundance with maintenance tobramycin therapy were facultative and obligate anaerobes; most are also known as oral microbiota constituents. These taxa have been associated with healthy airways,4 and their abundances have been noted to rise in CF respiratory samples prior to pulmonary exacerbations.37 38 Some of these taxa have also been implicated in worsening CF lung disease,39 40 and a recent study found that total bacterial load in bronchoalveolar lavage samples from children with CF correlated better with measures of structural lung disease than did the aggregate pathogen load, suggesting measuring all bacteria may be more clinically informative than considering just traditional pathogens.41 It is especially interesting to note that both the relative and absolute abundances of most anaerobic taxa we identified decreased after 1 week of therapy, rather than simply rising in relative abundance as those of dominant taxa decrease. Aminoglycosides, including tobramycin, have traditionally been thought to primarily impact aerobic taxa, as aerobic respiration efficiently drives electron transport-dependent aminoglycoside uptake.42 However, prior work has identified activity of aminoglycosides, although limited, under low-oxygen conditions,43 44 which are known to occur in CF secretions.37 Our data do not distinguish whether tobramycin had direct antimicrobial effects on specific taxa, as opposed to indirect effects through other, interacting microbes. If tobramycin is able to directly target these lower-abundance taxa, it is unclear from our data how these effects are related, if at all, to the long-term morbidity and mortality benefits of this common therapy. It is possible that such effects are directly related to the pathogenesis of CF lung disease, that reducing low-abundance taxon cell numbers has secondary effects on canonical pathogen behaviour with downstream ramifications for pathogenesis, or that these effects are entirely unrelated to clinical benefit. Regardless, the clinical consequences—both short-term and long-term—of these unintended effects require further study in order to translate these findings into clinical practice.
Our work has a number of limitations. Although we did detect a number of DNA viruses (primarily phage) and fungi (primarily Candida and Aspergillus), our DNA extraction method was not optimised to detect either of these kingdoms, and our sequencing platform cannot detect RNA viruses. Therefore, it is likely we did not reliably detect these taxa in our samples. However, given the relatively high abundances of bacteria in CF sputum, and the antibacterial effect of tobramycin, our analysis was intended to focus on bacteria. In addition, any bioinformatic method for taxonomic classification is limited by the fidelity of its database. Further work should be done to verify the species identities of the taxa we found to be significantly different between baseline and week 1 samples, particularly those which require more thorough genome sequencing to differentiate, such as the S. mitis, S. oralis, and S. pneumoniae group identified in figure 2B.45 Sputum variably samples the entire respiratory tract (including the oral cavity and upper respiratory tract). There is debate regarding how well sputum reflects lower airway microbiota, whether sputum always samples the same anatomic locations,5 and the extent to which oral bacteria are introduced to sputum during either aspiration or expectoration.46–48 Tobramycin concentrations in the oral cavity are likely high with inhaled therapy, with predicted effects on the oropharyngeal microbiome. Because this study was not designed to answer these questions, we report our results with respect to sputum microbiomes, without inferring relationships with lower airways. Samples were stored for 2–48 hours at 4°C prior to processing and long-term storage at −80°C, a commonly used approach we chose to maximise subject participation.41 Prior studies have indicated minimal changes in viable counts of P. aeruginosa49 or microbiota composition50 with extended storage at 4°C. While alterations in taxonomic or functional constituency of our samples under our storage conditions remain a possibility, because all samples were treated equally, any such changes are unlikely to have affected differences between baseline and week 1 samples. Further studies may benefit from immediate sample processing after collection. Furthermore, all subjects were clinically stable during the study, and the majority (24/30) were not naive to tobramycin maintenance. Of the 30 participants recruited to the study, 4 did not complete the study, of whom two experienced increased symptoms requiring alternative antibiotic therapy after week 1 and before week 4, and all samples collected after these new antibiotics were excluded (however, all 30 subjects had baseline and week 1 samples analysed). We chose these exclusion criteria to ensure all participants were clinically stable at the time of sample collection (limiting potential microbiota changes not attributable to antibiotic therapy). These exclusions would be expected to spuriously skew the clinical response to tobramycin positively (ie, by removing individuals who worsened during the study, we artificially enriched for individuals who had minimal or positive clinical response to tobramycin), which in turn may have further limited our power to detect microbial metrics associated with differential clinical response. Therefore, results may not reflect the effects of tobramycin on respiratory sample microbiomes from people during initial treatment or at later stages of disease. The work by Heirali et al therefore complements our study by focusing on older patients with little or no prior tobramycin exposure.
Our study also had notable strengths compared with prior work in this area. All participants were on only tobramycin, apart from maintenance azithromycin, for the duration of the study. Samples and data were collected at standardised time points with respect to treatment period, with weekly sampling during treatment. Variation in treatment and collection procedures were significant limitations of previous studies and may have limited resolution,5 39 51–53 and the relative homogeneity of our study design likely augmented our discriminatory power. While this focus may limit generalisability to other antibiotics or diseases, tobramycin is the antibiotic used most often to treat P. aeruginosa infections in PWCF, representing a large population of affected patients. Our results indicate the power of using standardised study designs to identify significant microbiome changes with treatment despite interpatient variability.
Our data complement previous studies that characterised the CF sputum microbiota via metagenomic sequencing, identifying diverse taxa not detected by classic clinical culture, and marked intersubject sputum microbial diversity.22 54–57 Moran Losada et al54 and Feigelman et al56 used metagenomic sequencing to investigate clonality of dominant pathogens and identified low-abundance strains in the CF sputum metagenome, in contrast to our findings. This difference may be due to the higher-stringency StrainPhlAn2 pipeline that we utilised to determine strain-level variability compared with whole-genome reconstruction and analysis used in prior work. Quinn et al characterised the functional capacity encoded by the CF sputum metagenome in 19 individuals with the specific goal of identifying keystone functional groups and their relationship to taxonomic composition.55 In contrast, we used metagenomic sequencing to specifically define the CF sputum microbial functional genomic capacities for a large number of samples and subjects over a single maintenance course of inhaled tobramycin, providing a rich view of the effects of tobramycin and identifying significant similarities across samples and with antibiotic perturbation. This analysis suggested a set of genes ‘necessary for life’ in CF respiratory secretions, similar to observations of other human-associated microbiomes.58 59 We also identified significant treatment-emergent abundance changes in genes related to antibiotic resistance and stress responses and carried largely by non-dominant taxa, perhaps underlying the well-known resilience of CF respiratory microbiota to therapy5 53 60 and suggesting a potential role for tobramycin therapy for a broader population of PWCF and related infections. While our study did not identify the mechanisms of the clinical effects of maintenance inhaled tobramycin, our data showed the largest microbiological impact of this therapy to be on non-dominant taxa, suggesting a causal role for antibiotics for the decreasing microbial diversity that has been observed in CF respiratory samples with increasing age, disease severity and antibiotic exposure.24 39 These findings indicate the power of metagenomic sequencing during treatments such as antibiotics, anti-inflammatories and cystic fibrosis transmembrane conductance regulator (CFTR) modulators to better define the microbial determinants of CF lung disease and treatment responses.
Methods
Study design and sample processing
The TIP study population comprised 30 PWCF at three clinical sites: University of Washington Medical Center, Seattle Children’s Hospital and University of Michigan Health System. Spontaneously expectorated sputum was collected prospectively at commencement of TIP (‘baseline’) and weekly during the month-long treatment period and shipped on ice within 48 hours of collection.50 Full details regarding the inclusion and exclusion criteria, clinical metrics, sample processing, culturing, DNA extraction and microbiome analysis of these specimens are provided in online supplementary methods.
Statistical analysis
For a priori analysis of all community characteristics to therapeutic response, we calculated change in ppFEV1 and CRISS symptom score from baseline to week 4 and compared with microbial metrics individually. A Benjamini-Hochberg correction for multiple comparisons was performed and p values smaller than 0.05 were considered significant. Further details regarding statistical analyses are provided in online supplementary methods.
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The accession number for all metagenomic sequencing and 16S amplicon sequencing data reported in this paper is NCBI Bioproject: PRJNA530252.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Dr Pradeep Singh for help with initial study design and thoughtful critique of this manuscript. We thank all people with cystic fibrosis who participated in this study.
References
Footnotes
Contributors MTN, JJL, RHS, DJW and LRH performed conceptualisation and design of the study. MTN, DJW, ATV, SR, GB, AR, MB, SN, LN and CM helped in data collection. MTN, AE, EJW, MJB, EB and LRH performed the data analysis and interpretation. Resources were collected by MJB, HSH, SIM, MB, SN, LN and CM. MTN and LRH helped in writing the original draft. All authors helped in writing, review and editing of the article. MTN helped in visualisation, DJW, EB, JJL, RHS and LRH helped in supervision and LRH, JJL and RHS helped in acquisition of funds.
Funding This work was supported by grants from the NIH (DK089507), from the Cystic Fibrosis Foundation (SINGH15R0), an institutional training grant (T32AI55396), and an unrestricted grant from Novartis.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.