Article Text

Abstract
Background Type 2 immune dysfunction contributes to acute lung injury and lethality following haemorrhagic shock (HS) and trauma. Group 2 innate lymphoid cells (ILC2s) play a significant role in the regulation of type 2 immune responses. However, the role of ILC2 in post-HS acute lung injury and the underlying mechanism has not yet been elucidated.
Objective To investigate the regulatory role of ILC2s in HS-induced acute lung injury and the underlying mechanism in patients and animal model.
Methods Circulating markers of type 2 immune responses in patients with HS and healthy controls were characterised. Using a murine model of HS, the role of high-mobility group box 1 (HMGB1)-receptor for advanced glycation end products (RAGE) signalling in regulation of ILC2 proliferation, survival and function was determined. And the role of ILC2 in inducing type 2 immune dysfunction was assessed as well.
Results The number of ILC2s was significantly increased in the circulation of patients with HS that was correlated with the increase in the markers of type 2 immune responses in the patients. Animal studies showed that HMGB1 acted via RAGE to induce ILC2 accumulation in the lungs by promoting ILC2 proliferation and decreasing ILC2 death. The expansion of ILC2s resulted in type 2 cytokines secretion and eosinophil infiltration in the lungs, both of which contributed to lung injury after HS.
Conclusions These results indicate that HMGB1-RAGE signalling plays a critical role in regulating ILC2 biological function that aggravates type 2 lung inflammation following HS.
- critical care
- innate immunity
- ARDS
- cytokine biology
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Type 2 immunity dysfunction is associated with haemorrhagic shock (HS)-induced acute lung injury and mortality; however, the underlying mechanism of type 2 immunity activation in the lungs following HS remains unclear.
What is the bottom line?
This study reveals a previously unidentified mechanism that high-mobility group box 1 (HMGB1) released during HS signals via receptor for advanced glycation end products (RAGE) to induce type 2 innate lymphoid cell (ILC2) expansion, with subsequent augments type 2 inflammation and eosinophil infiltration in the lungs, both of which contribute to HS-induced lung injury (Graphical Abstract).
Why read on?
This is the first report to show that HMGB1-RAGE-dependent expansion of ILC2s in the lungs displays an essential role in the development of type 2 immune dysfunction, suggesting that targeting HMGB1-RAGE signalling and therefore ILC2 expansion may present a novel preventive and therapeutic strategy for type 2 inflammation in lung injury.
Introduction
Haemorrhagic shock (HS), a result of major trauma or surgery, is one of the global leading causes of death with an estimated worldwide mortality of 1.9 million per year.1 Mortality in HS is largely due to the development of multiple organ dysfunction syndrome, of which acute lung injury (ALI) is an important component. HS promotes the development of ALI by exaggerating inflammation and subsequent immunosuppression, which causes nosocomial infections in the lungs and secondary complications, ultimately leads to organ dysfunction and mortality.2
Extensive studies addressed the important roles of type 1 immunity in the development of post-HS systemic inflammatory response syndrome.3 Recently, emerging data highlight the effect of type 2 immunity in the regulation of inflammation following tissue damage and infection.4 It is evident that patients with severe trauma have increased circulating levels of type 2 cytokines, including interleukin (IL)−4, IL-5 and IL-13 within 24 hours of injury.5 And the levels of type 2 cytokines are suggested to be correlated with an immunosuppressed status and nosocomial infections after injury.6 7 Traditional type 2 cytokines are mainly secreted by Th2 lymphocytes.8 Recent studies have shown that group 2 innate lymphoid cells (ILC2s) play significant role in the regulation of type 2 immunity9 10 However, the regulation mechanisms of ILC2s following HS are poorly elucidated.
High-mobility group box 1 (HMGB1) is a highly conserved non-histone architectural DNA-binding nuclear protein acting as a damage-associated molecular pattern (DAMP) molecule when released by dying cells or actively secreted by stressed cells.11 The receptors of HMGB1 include advanced glycation end products (RAGE) and other pattern recognition receptors such as TLR4 and TLR9.12 Studies have shown that different lymphoid cells including natural killer cells, B cells and T cells can express HMGB1 receptors. After HMGB1 binding to these receptors, it can promote the expansion and survival of CD4+ T-cells and CD8+ T-cells,13 induce the proliferation and activation of Th17 cells,14 and stimulate survival and migration of regulatory T cells in a RAGE-dependent fashion.15 16 Nevertheless, the role of HMGB1 signalling in homeostasis and function of ILC2s remains unknown.
Here, we show that the number of ILC2s in the circulation of patients with HS is significantly increased, and the increase in ILC2s is correlated with the recruitment of eosinophils and the plasma levels of type 2 cytokines IL-5 and IL-13 in patients with HS. Mechanistic studies reveal that HMGB1 released during HS induces lung ILC2 expansion in a RAGE-dependent manner through increasing ILC2 proliferation and decreasing ILC2 death. Expansion of ILC2s, in turn, results in enhanced type 2 inflammatory response and eosinophil infiltration in the lungs, followed by augmented lung injury after HS. These data identify HMGB1-RAGE as a novel regulator of ILC2s promoting type 2 inflammation in the lungs following HS.
Materials and methods
Additional methods are presented in the online supplementary file.
Supplemental material
Human subjects
Informed consent was obtained from all subjects or their surrogates. Totally, 34 patients with HS were enrolled from October 2018 to September 2019. Patients were included if they met the criteria for HS (systolic blood pressure ≤90 mm Hg and heart rate >108 beats/min or systolic blood pressure <70 mm Hg thought to be due to acute blood loss). Exclusion criteria included the following: age younger than 18 years or older than 90 years, shock with any other cause (cardiogenic shock or obstructive shock), or inability to provide informed consent. Clinical and demographic data were recorded within the first 24 hours after diagnosis of HS by two senior intensivists. Thirty-three healthy volunteers were recruited as controls.
HS mouse model
A mouse HS and resuscitation model was created as described previously.7 17 In brief, mice were anaesthetised with intraperitoneal injection (i.p.) of ketamine and xylazine. Femoral arteries were cannulated for monitoring of mean arterial pressure, blood withdrawal and fluid resuscitation. HS was then initiated by blood withdrawal and reduction of mean arterial pressure to 28–32 mm Hg. After a hypotensive period of 90 min, animals were resuscitated by transfusion of the shed blood and saline in a volume of equal to that of the shed blood. Sham animals underwent the same surgical procedures without haemorrhage and resuscitation. In some experiments, mice received neutralising antibody against HMGB1 (2 mg/kg, IBL International, Japan) or control non-specific chicken IgY (2 mg/kg, IBL International) by i.p. 30 min before haemorrhage.
Statistical analysis
Unpaired two-tailed Student’s t-test was used to quantify statistical deviation between different groups in human experiments. Correlations were analysed by using Spearman’s correlation coefficient. Mann-Whitney or Kruskal-Wallis test was used to reanalyse the data of mouse experiments. Statistical analyses were performed with SPSS V.21.0 (SPSS) or GraphPad Prism V.6.0 (GraphPad Software). Data are expressed as mean±SD. For all statistical analysis, p<0.05 was considered statistically significant.
Results
ILC2s and type 2 cytokines are elevated in the circulation of patients with HS
Thirty-four patients with HS and 33 healthy controls were enrolled and peripheral blood ILC2s and markers of type 2 responses were measured by flow cytometry and ELISA, respectively. The demographic and clinical characteristics of the subjects were listed in online supplementary table S1. The percentages and absolute numbers of peripheral blood ILC2s were significantly increased in patients 24 hours after HS, as compared with those in healthy individuals (figure 1A). Similarly, the percentages of peripheral blood eosinophils were also significantly higher in patients with HS as compared with control subjects, but the proportions of neutrophils did not change significantly in patients with HS (figure 1B). Furthermore, plasma concentrations of IL-5 and IL-13 were also markedly increased at 24 hours after HS (figure 1C) while plasma concentrations of IL-4 and IL-9 did not show statistic differences between the groups of HS and healthy controls (data not shown). Consistently, HS induced increase in the percentages of IL-5+ ILC2s and IL-13+ ILC2s in peripheral blood (online supplementary figure 1)
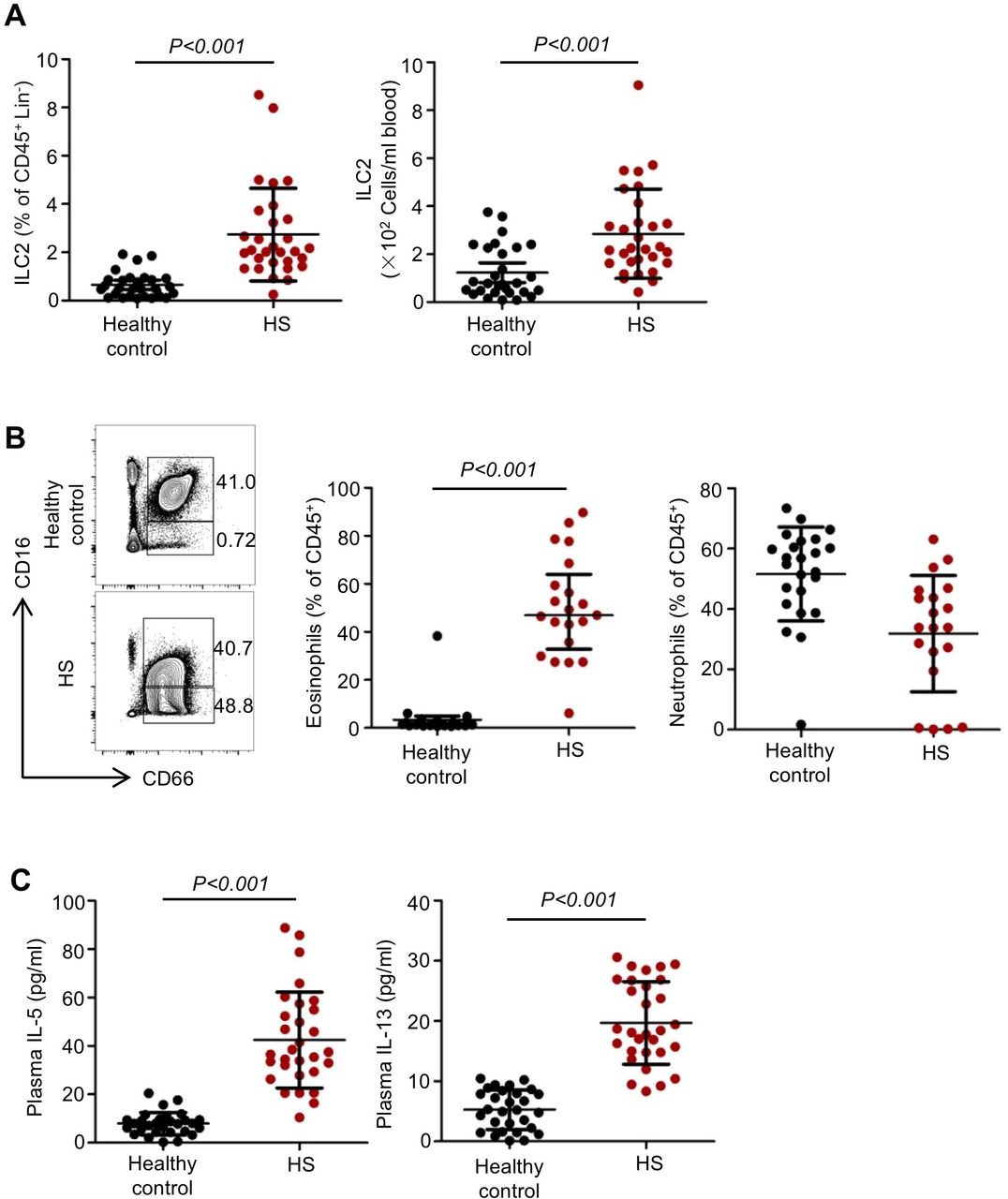
Elevated circulating type 2 innate lymphoid cells (ILC2s) and type 2 immune responses in patients with haemorrhagic shock (HS). (A) Graphs showing ILC2 percentages within the CD45+ Lin− populations and absolute numbers of ILC2s in the peripheral blood from the studied subjects. n=29 for healthy controls, n=30 for patients with HS. (B) Representative flow cytometric analysis and graphs showing percentages and absolute numbers of eosinophils (CD45+ CD16− CD66+ CD14−) and neutrophils (CD45+ CD16+ CD66+ CD14−) within the CD45+ populations in the blood from the studied subjects. n=24 for healthy controls, n=20 for patients with HS. (C) ELISA analysis of plasma interleukin (IL)-5 and IL-13 concentrations in 30 patients with HS and 29 healthy controls. Dots represent individual subjects. All results are shown as mean±SD. Data were analysed by unpaired two-tailed Student’s t-test.
Expansion of peripheral ILC2s correlates with markers of type 2 immune responses and clinical complications
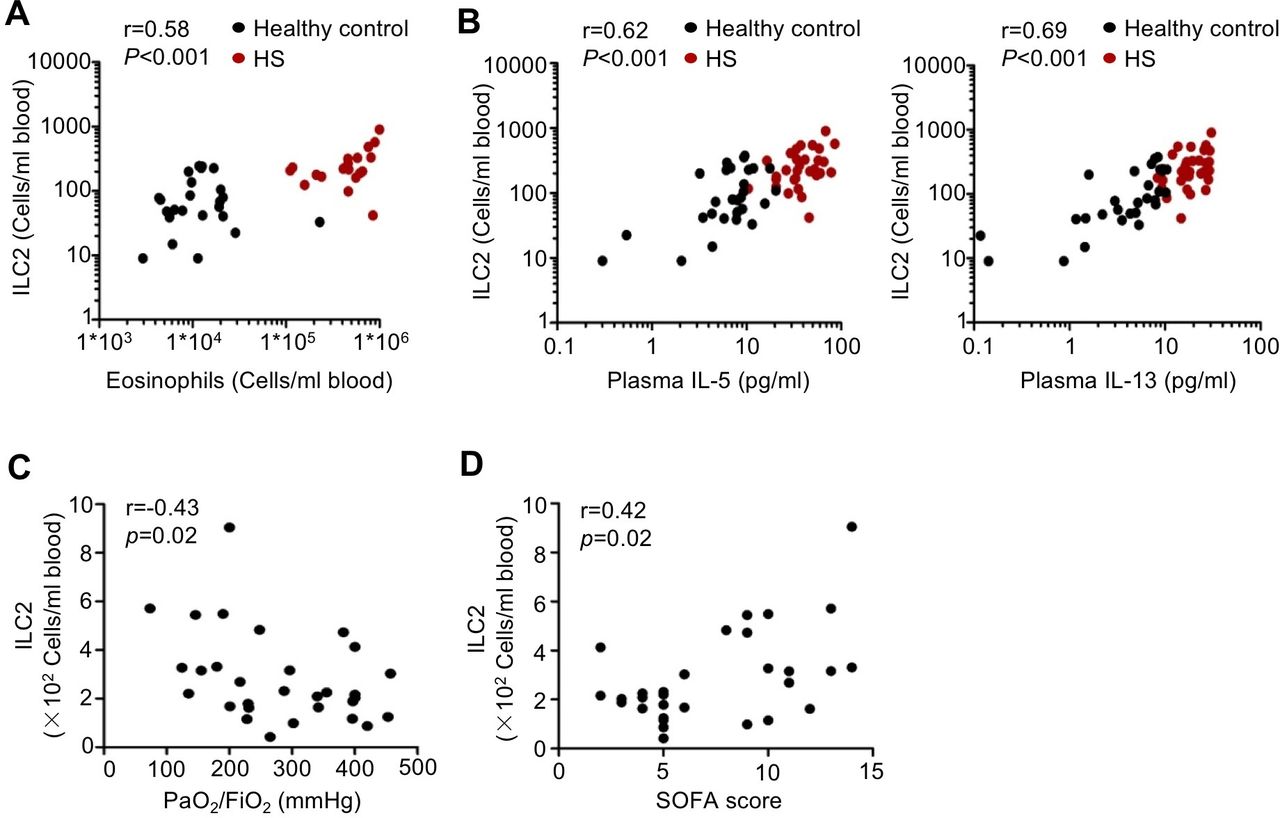
Next, we sought to determine whether expansion of ILC2s was associated with the amplification of circulating type 2 immune responses. The absolute numbers of peripheral blood ILC2s at 24 hours after HS were positively correlated with the numbers of peripheral blood eosinophils (figure 2A) and the levels of plasma IL-5 and IL-13 levels (figure 2B). We also explored the relationship between ILC2s and clinical complications of patients with HS. Interestingly, we found that circulating ILC2 numbers at 24 hours after HS were negatively correlated with PaO2/FiO2 ratio (figure 2C), and positively with Sequential Organ Failure Assessment score of the patients with HS (figure 2D). Taken together, the human studies show that peripheral blood ILC2 numbers are elevated in patients with HS and the increased numbers of ILC2s correlate with circulating type 2 cytokines levels and clinical complications.

Circulating type 2 innate lymphoid cells (ILC2s) correlate with markers of type 2 responses and clinical complications. (A) Correlation of peripheral blood ILC2s with peripheral blood eosinophil numbers 24 hours after haemorrhagic shock (HS) (n=24 for healthy controls, n=20 for patients with HS). (B) Correlation of peripheral blood ILC2s with plasma interleukin (IL)-5, IL-13 levels 24 hours after HS (n=29 for healthy controls, n=30 for patients with HS). (C) Correlation of blood ILC2 numbers 24 hours after HS with PaO2/FiO2 ratio in the patients with HS (n=30). (D) Correlation of blood ILC2 numbers 24 hours after HS with Sequential Organ Failure Assessment (SOFA) score in the patients with HS (n=30). Data are presented as Spearman’s rank correlation. Dots represent individual subjects.
HMGB1 induces ILC2 expansion following HS
To determine the role of ILC2 expansion in local type 2 immune responses, mouse HS model was used and ILC2 expansion in the lungs was dynamically measured by flow cytometry. The percentages and absolute numbers of ILC2s were significantly increased in the lungs by 6 hours, reached a peak at 24 hours and remained elevated for 48 hours after HS (online supplementary figure 2A,B).
Clinical study indicated that plasma concentrations of HMGB1 were elevated in patients with HS, and HMGB1 levels were significantly correlated with the number of peripheral blood ILC2s at 24 hours after HS (figure 3A). Next, we detected HMGB1 levels in the lungs and plasma in murine HS model, and found that HMGB1 levels increased by 6 hours, reached a peak at 24 hours and remained elevated for 48 hours in the lungs (figure 3B,C). Moreover, we found that HS-induced increase of HMGB1 in the lungs was mainly derived from infiltrated neutrophils and macrophages (online supplementary figure 3A,B).
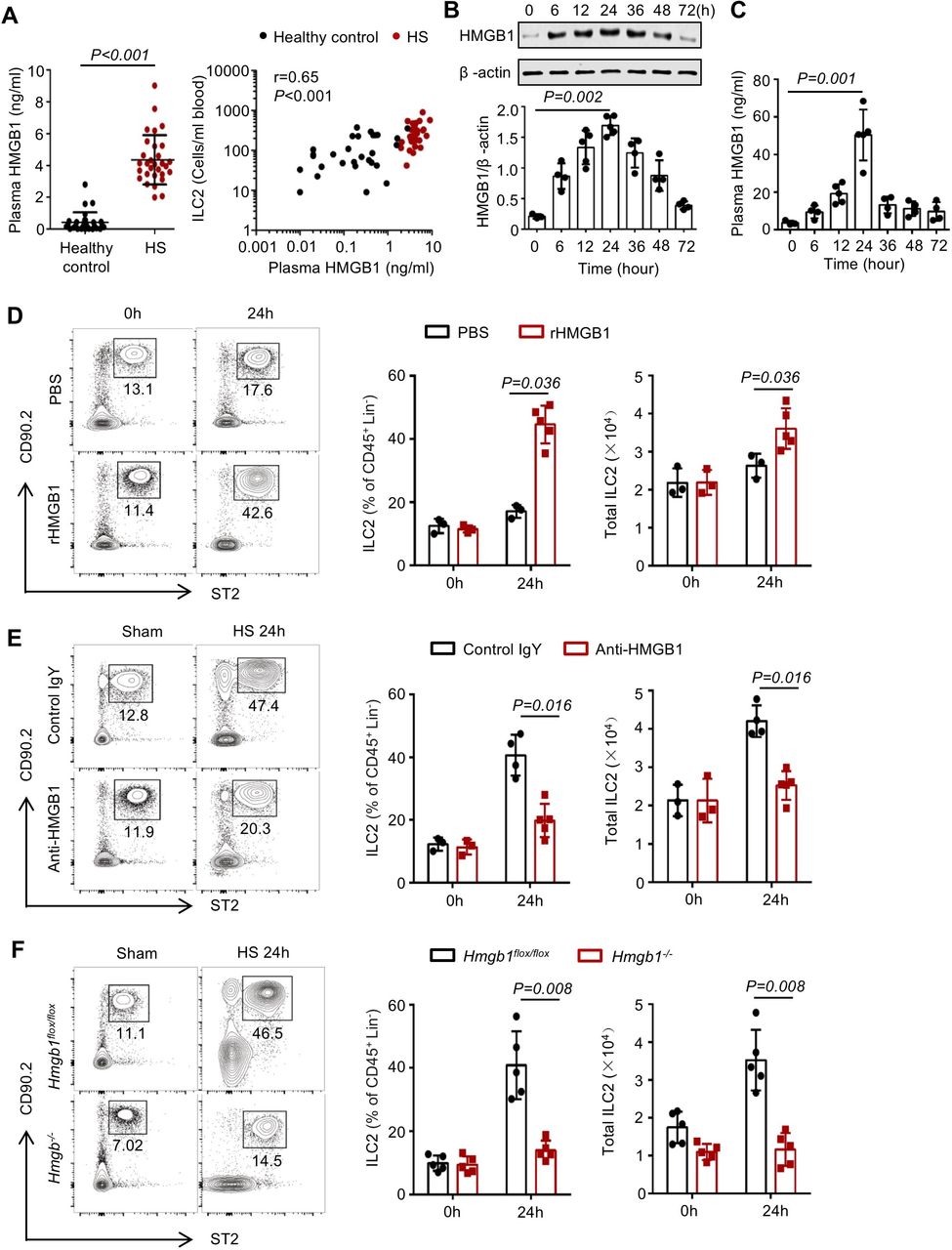
High-mobility group box 1 (HMGB1) induces type 2 innate lymphoid cells (ILC2s) accumulation in the lungs following haemorrhagic shock (HS). (A) ELISA analysis of plasma HMGB1 concentrations and correlation of plasma HMGB1evels with absolute numbers of peripheral blood ILC2s in 30 patients with HS and 29 healthy controls. (B) Western blot analysis of HMGB1 protein expression in lung tissues from wild-type (WT) mice at 0, 6, 12, 24, 36, 48 and 72 hours after HS or sham surgery. Graphs showing quantification of density of Western blot HMGB1 bands normalised to β-actin loading control. (C) ELISA analysis of plasma HMGB1 concentrations in WT mice at time points up to 72 hours after HS. (D) Representative flow cytometry plots, pregated for CD45+ Lin− cells, showing percentages of ILC2s in the lungs of WT mice given intratracheally rHMGB1 (2 mg/kg) or phosphate buffer saline (PBS) for 24 hours. Graphs showing percentages and total numbers of ILC2s in the lungs. (E) Representative flow cytometry plots showing ILC2 percentages and graphs showing ILC2 percentages within the CD45+ Lin− populations and absolute numbers of ILC2s in the lungs at 24 hours after HS or sham surgery in WT mice treated with intraperitoneal neutralising antibody against HMGB1 (anti-HMGB1, 2 mg/kg), or non-specific control antibody (control IgY). (F) Representative flow cytometry plots showing percentages of ILC2s within the CD45+ Lin− populations in the lungs and graphs showing ILC2 percentages and absolute numbers of ILC2s in the lungs of inducible Hmgb1−/− or control mice at 24 hours after HS. All results are shown as mean±SD. Data were analysed by (A) unpaired two-tailed Student’s t-test and Spearman’s rank correlation, (B, C) Kruskal-Wallis test or (D–F) Mann-Whitney test.
To determine whether extracellular HMGB1 mediates ILC2 expansion, we treated wild-type (WT) mice with rHMGB1 or PBS injected intratracheally (i.t.) and assessed ILC2 numbers in the lungs at 24 hours. Lung ILC2s were significantly increased by rHMGB1 i.t. as compared with the PBS controls (figure 3D). Next, we gave mice neutralising anti-HMGB1 antibody via i.p. 30 min before haemorrhage. Compared with control IgY-treated HS mice, anti-HMGB1 treatment groups had a significantly decreased number of ILC2s in the lungs (figure 3E). Furthermore, we found that HMGB1 deficiency prevented ILC2 expansion in the lungs at 24 hours following HS, and reduced ILC2s in the lungs at baseline (figure 3F).
HMGB1-mediated accumulation of lung ILC2s requires RAGE
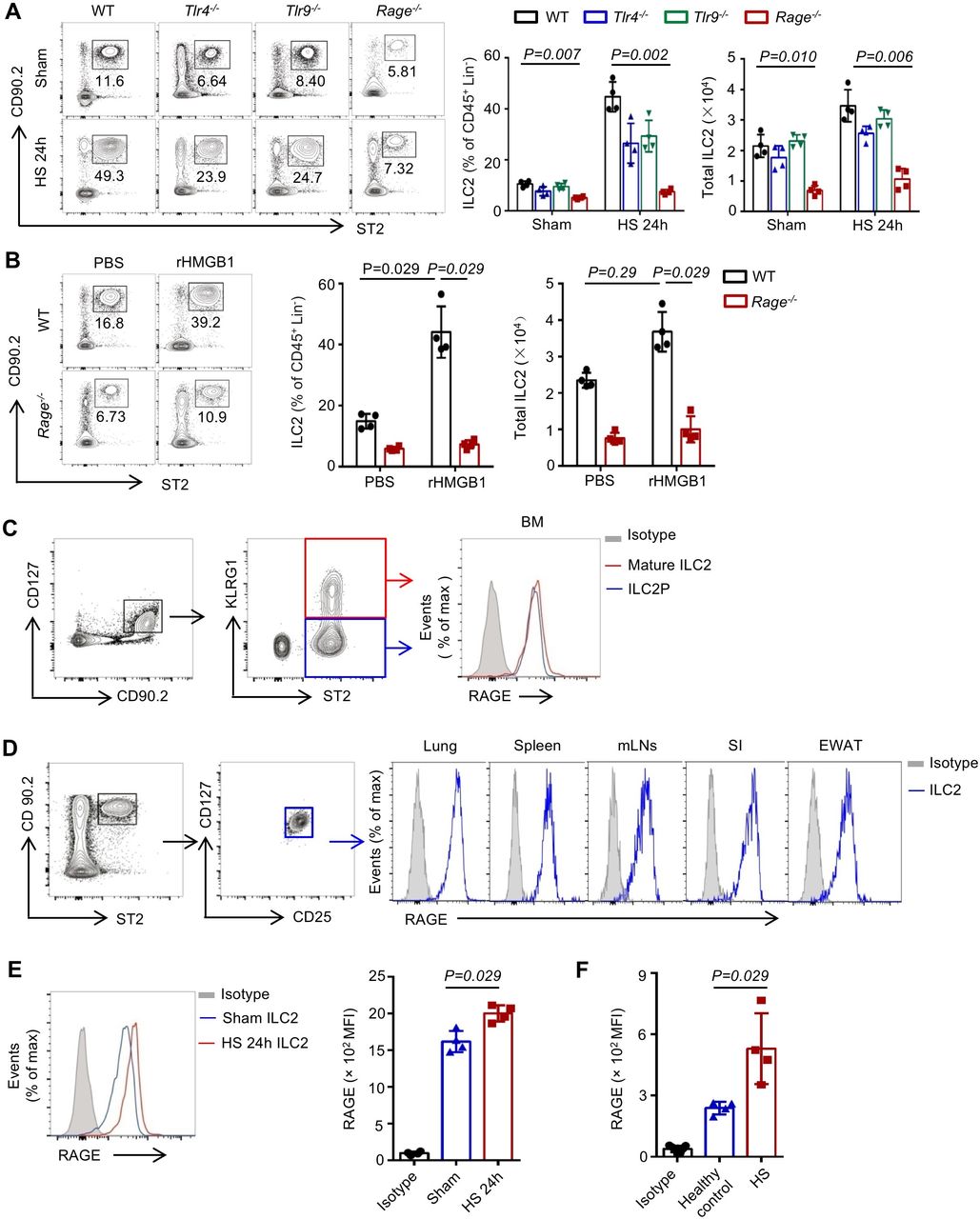
Receptors for HMGB1 include TLR4, TLR9 and RAGE.18 We therefore subjected Rage−/−, Tlr4−/− and Tlr9−/− mice to HS, and measured ILC2 numbers in the lungs at 24 hours. As shown in figure 4A, RAGE deficiency effectively prevented ILC2 expansion in the lungs following HS. Next, we gave WT or Rage−/− mice rHMGB1 i.t. and found that rHMGB1 failed to increase ILC2s in the lungs in Rage−/− mice at 24 hours after the injection (figure 4B).

High-mobility group box 1 (HMGB1) mediates type 2 innate lymphoid cells (ILC2s) expansion in the lungs through receptor for advanced glycation end products (RAGE). (A) Representative flow cytometry plots showing percentages of ILC2s within the CD45+ Lin− populations and graphs showing percentages and absolute numbers of ILC2s in the lungs of wild-type (WT), Tlr4−/−, Tlr9−/− and Rage−/− mice at 24 hours after HS or sham surgery. (B) Representative flow cytometry plots showing percentages of ILC2s within the CD45+ Lin− populations and graphs showing percentages and absolute numbers of ILC2s in the lungs of WT and Rage−/− mice at 24 hours after intratracheally instillation of rHMGB1 (2 mg/kg) or PBS. (C) Flow cytometric analysis of RAGE expression on bone marrow immature ILC2 precursors (CD45+ Lin− CD90.2+ CD127+ ST2+ KLRG1−) and mature ILC2s (CD45+ Lin− CD90.2+ CD127+ ST2+ KLRG1+) from WT mice. (D) Flow cytometric analysis of RAGE expression on mature ILC2s in lungs and spleen, mesenteric lymph nodes (mLNs), small intestine (SI) and epididymal white adipose tissue (EWAT) of WT mice. (E) Flow cytometric analysis of RAGE expression and graphs showing mean fluorescent intensity (MFI) levels of RAGE expression on mature lung ILC2s at 24 hours after HS or sham. (F) Graphs showing mean MFI levels of RAGE expression on human blood ILC2s. All results are shown as mean±SD. Data were analysed by (A) Kruskal-Wallis test or (B, E, F) Mann-Whitney test.
We detected high RAGE expression on both lineage-specified BM ILC2 precursors and mature ILC2s (figure 4C). We also observed that mature ILC2s expressed high levels of RAGE in the lungs, spleen, mediastinal lymph nodes, small intestine and epididymal white adipose tissue in WT mice (figure 4D), indicating that constitutive RAGE expression represents a conserved trait of ILC2s across lymphoid and non-lymphoid tissues. Moreover, we found that RAGE expression was increased in the lung ILC2s at 24 hours after HS (figure 4E). Interestingly, RAGE expressed in human blood ILC2s and the levels of RAGE increased in blood ILC2s of patient with HS (figure 4F). Collectively, these observations show that HMGB1-RAGE is the major signalling pathway regulating the homeostasis of ILC2 after HS.
HMGB1-RAGE regulates ILC2 proliferation and survival after HS
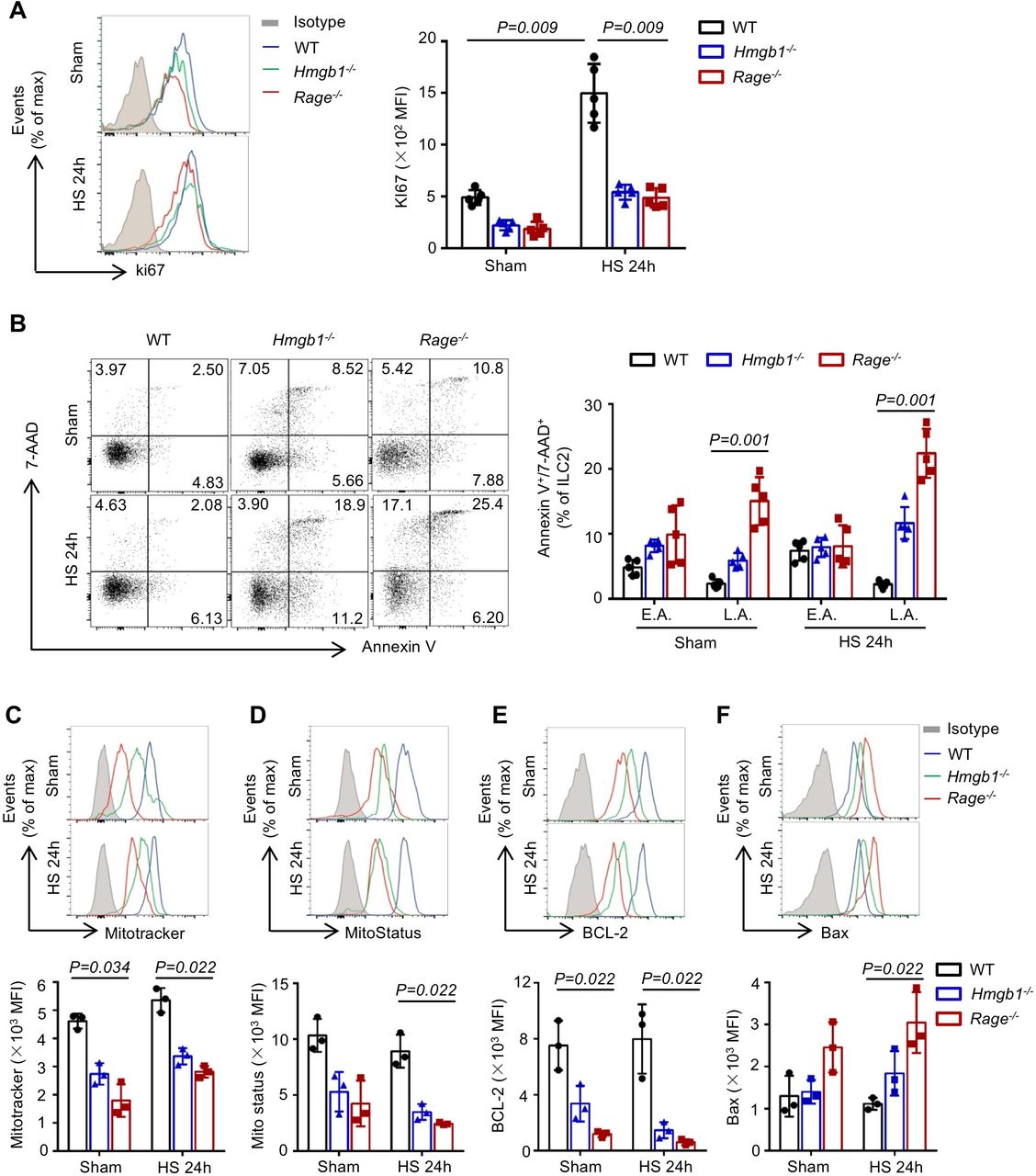
To elucidate the mechanisms of HMGB1-RAGE-mediated ILC2 expansion in the lungs following HS, we investigated the effect of HMGB1 or RAGE gene deletion on ILC2 proliferation, differentiation and survival. WT, Hmgb1−/− and Rage−/− mice were subjected to HS, and lung ILC2s were stained with Ki-67, a cell proliferation-associated nucleic protein that marks cells in active phases of the cell cycle. We detected high expression of Ki-67 in the ILC2s from lungs of WT mice at 24 hours after HS. In contrast, genetic deletion of HMGB1 or RAGE significantly reduced Ki-67 expression in ILC2s (figure 5A), indicating that HMGB1-RAGE signalling regulates the proliferation of lung ILC2s in response to HS.

High-mobility group box 1 (HMGB1)-receptor for advanced glycation end products (RAGE) regulates type 2 innate lymphoid cells (ILC2s) proliferation and survival after haemorrhagic shock (HS). (A) Representative flow cytometry histograms and graphs showing mean fluorescence intensity (MFI) levels of Ki-67 fluorescence in lung ILC2s (CD45+ Lin− CD90.2+ ST2+) from wild-type (WT), Hmgb1−/− and Rage−/− mice at 24 hours after HS or sham surgery. (B) Representative flow cytometry dot plots and graphs showing percentages of lung ILC2s staining positive for 7-amino-actinomycin D (7AAD) and Annexin V in WT, Hmgb1−/− and Rage−/− mice at 24 hours after HS. Early apoptotic ILC2s (E.A.; Annexin V+, 7AAD−), late apoptotic ILC2s (L.A.; Annexin V+, 7AAD+). Flow cytometric analysis and MFI levels of (C) Mitotracker, (D) MitoStatus, (E) BCL-2 and (F) Bax fluorescence in lung ILC2s from the same mice as above. All results are shown as mean±SD. Data were analysed by Kruskal-Wallis test.
GATA3 is an essential transcriptional factor for ILC2 differentiation and maintenance.19 20 Therefore, we determined GATA3 expression in lung ILC2s from WT, Hmgb1−/− and Rage−/− mice at 24 hours after HS. As shown in online supplementary figure 4A, HS significantly elevated GATA3 expression in ILC2s from WT, Hmgb1−/− and Rage−/− mice, but there was no obvious difference among these groups after HS. Interestingly, further analysis revealed that the proportions of immature ILC2s were significantly increased in the lungs of Hmgb1−/− and Rage−/− mice, as compared with WT, after HS (online supplementary figure 4B). These observations support that HMGB1-RAGE is not required for GATA3 expression in ILC2s after HS, but plays an essential role in the development of mature ILC2s in the lungs.
To test whether loss of HMGB1-RAGE affects ILC2 survival, WT, Hmgb1−/− and Rage−/− mice were subjected to HS. At 24 hours after HS, lungs ILC2s were double stained with 7-AAD and Annexin-V to determine cell death. As shown in figure 5B, genetic deficiency in HMGB1 or RAGE significantly increased ILC2 early apoptosis (Annexin-V+, 7-AAD−) and late apoptosis (Annexin-V+, 7-AAD+) in response to HS and even sham operation as compared with WT.
HMGB1-RAGE signalling can promote cell survival by regulating mitochondrial function.21 22 Lung ILC2s from WT, Hmgb1−/−, and Rage−/− mice at 24 hours after HS were stained with MitoTracker and MitoStatus Red to detect mitochondrial mass and depolarisation. We found that mitochondrial mass (figure 5C) and mitochondrial membrane potential (figure 5D) in ILC2s from Hmgb1−/− and Rage−/− mice were decreased as compared with that in WT mice following sham or HS. The ratio of Bcl-2 and Bax determines mitochondrial outer membrane permeabilisation, which often correlates with the loss of inner mitochondrial membrane potential.23 Next, we determined the expression of Bcl-2 and Bax in lung ILC2s at 24 hours after HS. Bcl-2 expression in ILC2s from Hmgb1−/− or Rage−/− mice was significantly suppressed compared with WT mice (figure 5E). However, Bax expression in ILC2s was increased in Rage−/− mice in response to HS (figure 5F). Collectively, these studies demonstrate that HMGB1-RAGE signalling promotes ILC2 survival through attenuating mitochondrial-mediated apoptosis.
Loss of HMGB1-RAGE signaling attenuates ILC2-induced type 2 inflammation
ILC2s are an important cellular source of type 2 cytokines.4 24 To determine whether HMGB1 contributes to ILC2 expansion induced type 2 inflammation in the lungs, we gave WT mice rHMGB1 i.t. and found HMGB1 treatment significantly increased IL-5+ ILC2s, IL-9+ ILC2s and IL-13+ ILC2s in the lungs (online supplementary figure 5A-C) but did not increase IL-4+ ILC2s (data not shown). Consistently, HS induced increase in the percentages of IL-5+ ILC2s, IL-9+ ILC2s and IL-13+ ILC2s in WT mice (figure 6A–C). However, genetic deletion of HMGB1 or RAGE attenuated HS-induced increase in IL-5+ ILC2s, IL-9+ ILC2s and IL-13+ ILC2s (figure 6A–C). Next, blocking of HMGB1 also led to significantly decrease of IL-5+ ILC2s, IL-9+ ILC2s and IL-13+ ILC2s at 24 hours after HS (figure 6D–F).

High-mobility group box 1 (HMGB1)-receptor for advanced glycation end products (RAGE) is required for type 2 innate lymphoid cell (ILC2)-induced type 2 inflammation. (A–C) Flow cytometry blots and graphs showing percentages of (A) IL-5+ ILC2s, (B) IL-9+ ILC2s and (C) IL-13+ ILC2s in the lungs of wild type (WT), Hmgb1−/− and Rage−/− mice at 24 hours after haemorrhagic shock (HS) or sham surgery. (D–F) Flow cytometry blots and graphs showing percentages of (D) interleukin (IL)-5+ ILC2s, (E) IL-9+ ILC2s and (F) IL-13+ ILC2s in the lungs at 24 hours after HS or sham surgery in WT mice treatment with HMGB1 neutralising antibody (2 mg/kg) or non-specific control antibody (control IgY). (G) ELISA analysis of plasma IL-5, IL-9 and IL-13 concentration at 24 hours after HS or sham surgery in WT, Hmgb1−/− and Rage−/− mice. (H) ELISA analysis of plasma IL-5, IL-9 and IL-13 concentration at 24 hours after HS or sham surgery in WT mice treated with HMGB1-neutralising antibody or control IgY. All results are shown as mean±SD. Data were analysed by Mann-Whitney test.
Plasma concentrations of type 2 cytokines were also measured following rHMGB1 i.t. or HS. IL-5, IL-9 and IL-13 in plasma were markedly increased at 24 hours after rHMGB1 treatment (online supplementary figure 5D) or HS (figure 6G). As expected, genetic deletion of HMGB1 or RAGE reduced IL-5, IL-9 and IL-13 in plasma after HS as compared with WT mice (figure 6G). In addition, blocking of HMGB1 suppressed IL-5, IL-9 and IL-13 production in plasma at 24 hours post-HS (figure 6H). Furthermore, ILC2-depletion experiments were carried out.25 Treatment of Rag1−/− mice with anti-Thy1.2 antibody effectively depleted ILC2 numbers in the lungs after rHMGB1 i.t (online supplementary figure 6A). Depletion of ILC2s attenuated rHMGB1-induced type 2 inflammation in the lungs (online supplementary figure 6B). Taken together, these data indicate type 2 cytokines produced by ILC2s are dependent HMGB1-RAGE specific pattern in the lungs after HS.
HMGB1-RAGE-dependent ILC2s promote eosinophil infiltration in the lungs
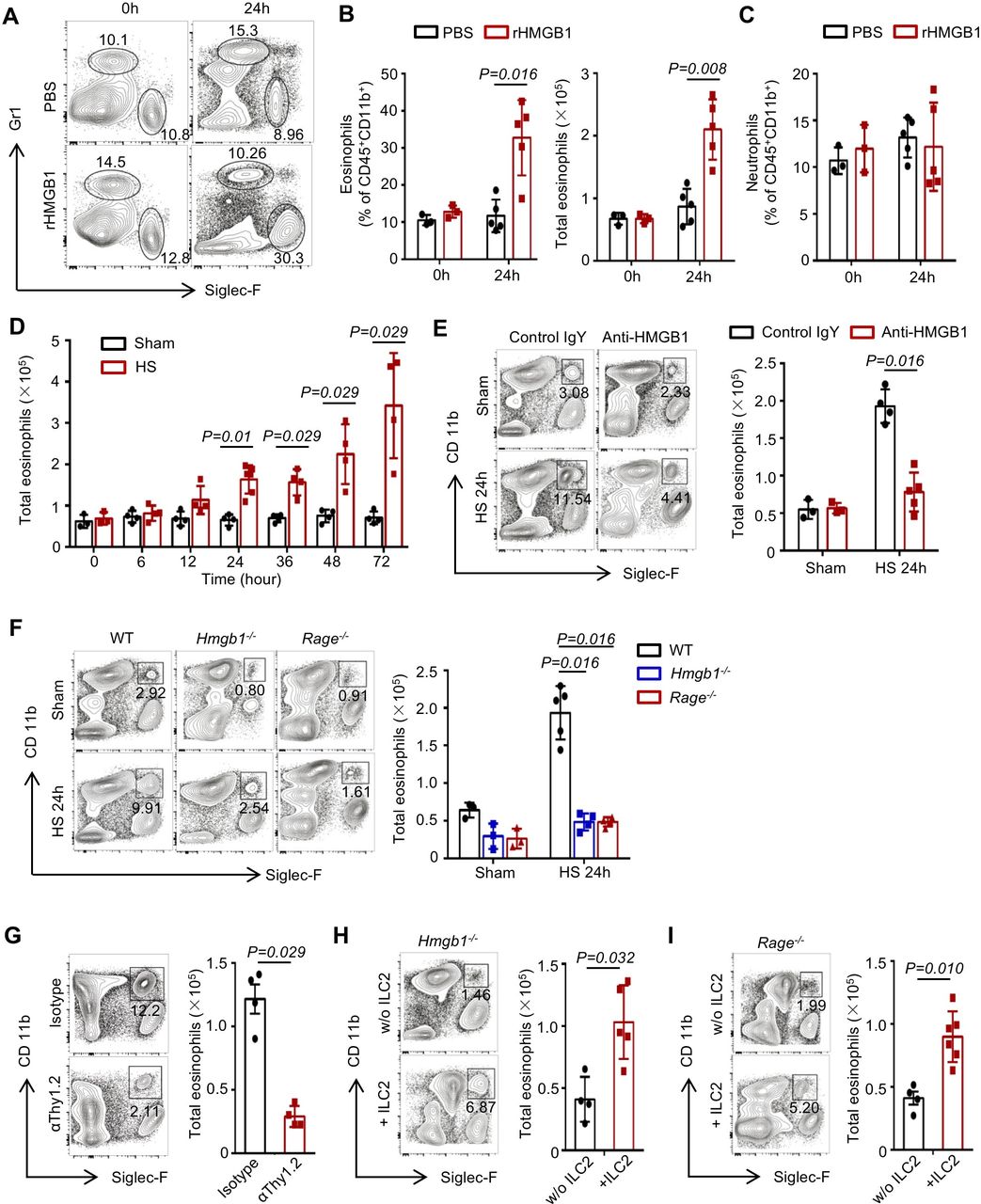
ILC2-derived IL-5, IL-9 and IL-13 are associated with eosinophil recruitment and survival in the lungs.26–28 We observed that at 24 hours after rHMGB1 i.t. to WT mice, eosinophils in the lungs were significantly increased compared with the PBS-treated controls (figure 7A,B). However, rHMGB1 did not induce a significant increase of neutrophils (figure 7C). Furthermore, time-course studies showed that amplification of eosinophils in the lungs after HS was consistent with the expansion of ILC2s (figure 7D). Next, blocking of HMGB1 led to significantly decrease of eosinophils at 24 hours post-HS (figure 7E). Genetic deletion of HMGB1 or RAGE mice also suppressed HS-induced eosinophil recruitment in the lungs (figure 7F).
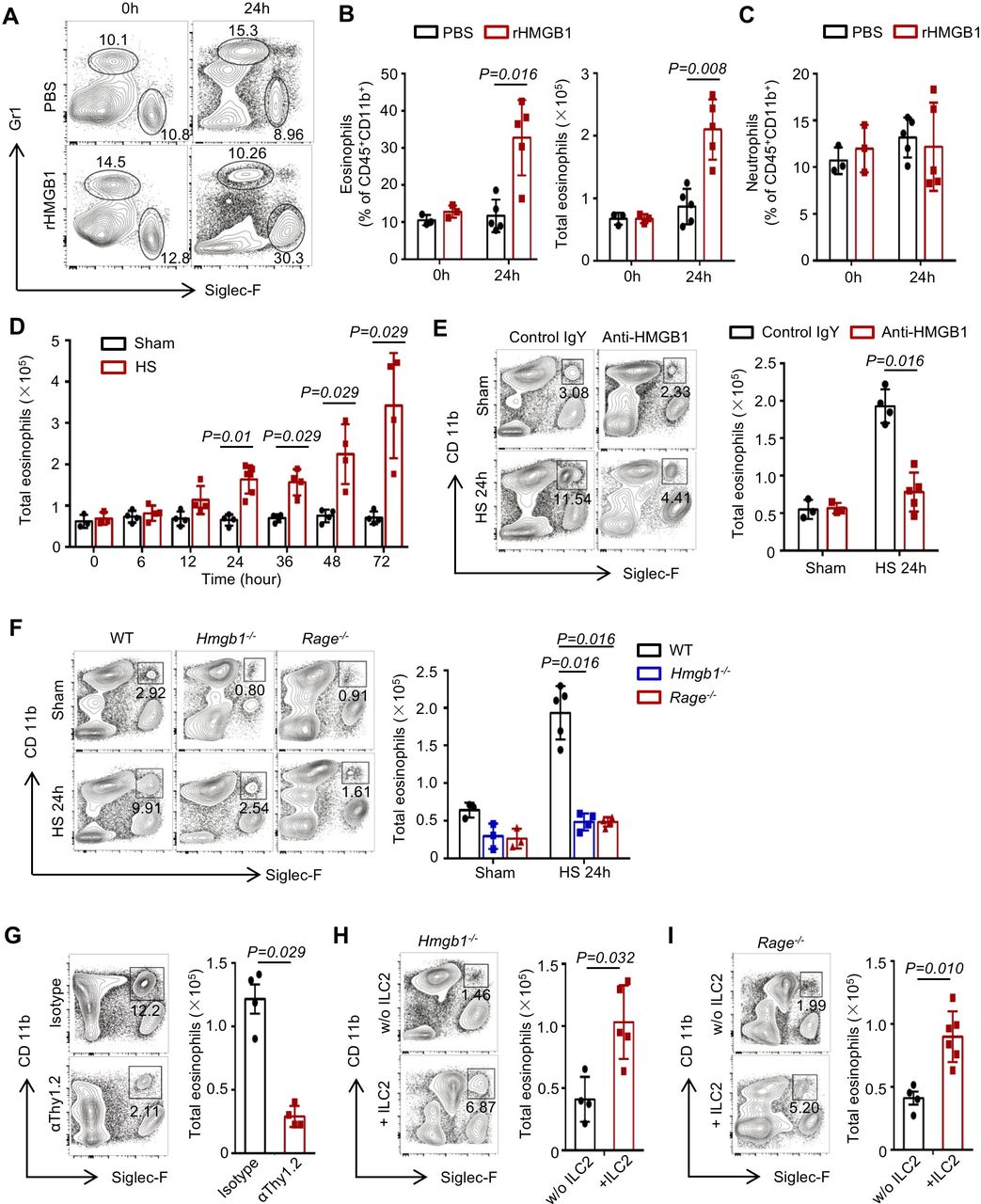
High-mobility group box 1 (HMGB1)-receptor for advanced glycation end products (RAGE) activation on innate lymphoid cells (ILC2s) induces eosinophil infiltration into lung. (A) Representative flow cytometric analysis and graphs showing (B) percentages and absolute numbers of eosinophils (CD45+ Gr-1− Siglec-F+ CD11b+) and (C) percentages of neutrophils (CD45+ Gr-1+ CD11b+ Siglec-F−) in the lungs of wild-type (WT) mice at 24 hours after treatment with intratracheally (i.t.) instilled rHMGB1 (2 mg/kg) or PBS. (D) Graphs showing absolute numbers of eosinophils measured by flow cytometry in the lungs of WT mice at time points up to 72 hours after haemorrhagic shock (HS). (E) Representative flow cytometry blots showing percentages within the CD45+ populations and graphs showing total number of eosinophils at 24 hours after HS in the lungs of WT mice treated with HMGB1-neutralising antibody or control IgY. (F) Representative flow cytometry plots showing percentages of eosinophils and graphs showing absolute numbers of ILC2s in the lungs of WT, Hmgb1−/− and Rage−/− mice at 24 hours after HS or sham surgery. (G) Flow cytometry plots showing percentages and graphs showing absolute numbers of lung eosinophils from isotype and αThy1.2-treated Rag1−/− mice 24 hours after given i.t. rHMGB1 gated on CD45+ populations. (H, I) Flow cytometry blots showing percentages and graphs showing absolute numbers of eosinophils in the lungs at 24 hours after HS in (H) Hmgb1−/− and (I) Rage−/− mice transferred with ILC2s (5×104 cells/mouse) isolated from WT mice or saline at 30 min post-HS. All results are shown as mean±SD. Data were analysed by Mann-Whitney test.
To directly establish a link between ILC2 expansion and eosinophil accumulation in the lungs following HS, we first confirmed that depletion of ILC2s attenuated rHMGB1-induced eosinophils increase (figure 7G). Then, we adoptively transferred ILC2s into Hmgb1−/− or Rage−/− mice. As shown in figure 7H,I, replenishing ILC2s in Hmgb1−/− or Rage−/− HS mice restored eosinophil infiltration in the lungs at 24 hours. Furthermore, we demonstrated that the ILC2s were recovered in the lung of HS-challenged Hmgb1−/− and Rage−/− mice by adoptive transfer of ILC2s (online supplementary figure 7A, B). C57BL/6J (CD45.1) mice were applied to test the transfer efficiency. We purified ILC2s from the lung of C57BL/6J (CD45.1) mice and intravenously injected into Rage−/− HS mice (CD45.2). The transfer efficiency revealed that the majority of ILC2s in the lung (78.94%±7.35%), mLNs (61.04%±9.62%) and spleen (85.78%±5.22%) were derived from CD45.1 mice (online supplementary figure 7C). These results suggest that HMGB1-RAGE signalling mediated ILC2 expansion may contribute to subsequent eosinophil infiltration in the lungs following HS.
HMGB1-RAGE-mediated ILC2 expansion aggravates lung injury after HS
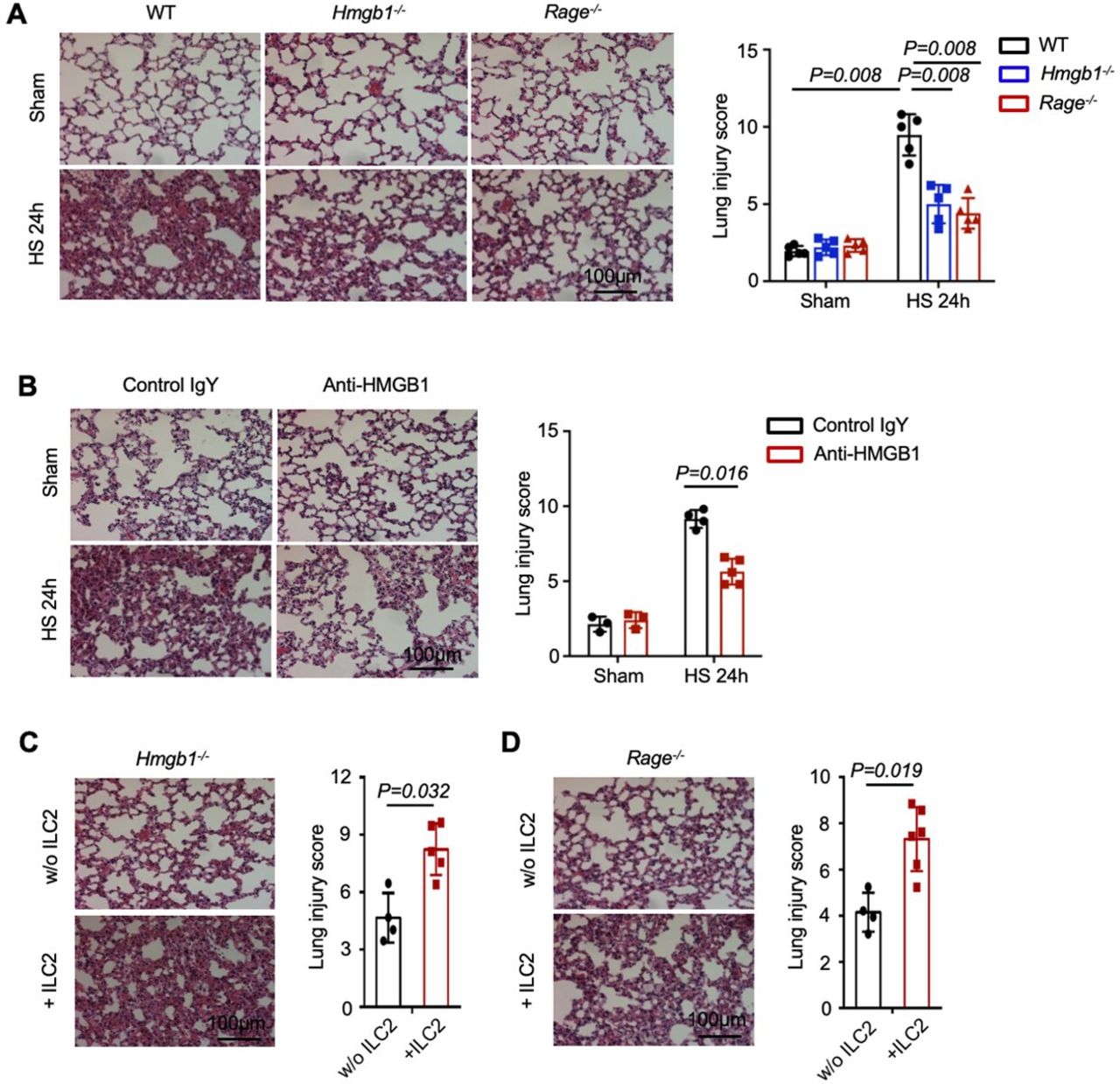
To address whether HMGB1-RAGE-mediated ILC2 expansion enhances lung injury, we assessed lung histology post-HS. Genetic deletion of HMGB1, RAGE or blocking of HMGB1 attenuated HS-induced lung injury (figure 8A,B). In addition, adoptively transferred WT ILC2s significantly worsened lung histological changes in response to HS in the Hmgb1−/− or Rage−/− mice (figure 8C,D). These data demonstrate a critical role of HMGB1-RAGE-dependent ILC2 expansion in promoting HS-induced lung injury.

High-mobility group box 1 (HMGB1)-receptor for advanced glycation end products (RAGE)-dependent type 2 innate lymphoid cells (ILC2s) expansion promotes lung injury after haemorrhagic shock (HS). (A) H&E stained sections and histopathological lung injury scores at 24 hours after HS of wild-type (WT), Hmgb1−/− and Rage−/− mice. (B) H&E stained sections and histopathological lung injury scores at 24 hours after HS from WT mice treated with HMGB1-neutralising antibody or control IgY. (C, D) H&E stained sections and histopathological lung injury scores at 24 hours after HS from (C) Hmgb1−/− and (D) Rage−/− mice transferred with ILC2s (5×104 cells/mouse) isolated from WT mice or saline at 30 min post-HS. Magnification is ×200. All results are shown as mean±SD. Data were analysed by Mann-Whitney test.
Discussion
Recently, emerging data highlighted the impact of exaggerated type 2 immunity following tissue damage and infection.4 The current study indicates that patients with HS present elevated ILC2s in the circulation and increased systemic type 2 inflammation. Moreover, mechanistic studies show that HMGB1-RAGE signalling promotes ILC2 proliferation and survival in the lungs after HS. Accumulation of ILC2s results in type 2 cytokine release and eosinophil infiltration in the lungs, both of which enhance post-HS ALI.
Type 2 immunity, which is characterised by elevations of IL-4, IL-5, IL-9 and IL-13, mediates host protection through reducing tissue inflammation and enhancing tissue repair. Nevertheless, when type 2 immune responses overexuberant, dysregulated, or become chronic, they can also contribute to the development of organ dysfunction.4 29 Recent studies showed that type 2 immune responses are one of the major drivers of the immune dysregulation and nosocomial infections following HS.6 Severe trauma patients with HS exhibit higher circulating levels of IL-33, which may lead to deleterious type 2 immune responses.7 This study, however, first demonstrates that HMGB1 initiates ILC2-driven type 2 immunity, which serves as a detrimental factor in the HS-induced lung injury.
We have previously demonstrated that sepsis-induced ILC2 accumulation in the lungs protects lung endothelial cells from pyroptosis.30 However, the consequences of ILC2 expansion in the development of lung injury are controversial. Some studies show that ILC2 activation contributes to inflammation associated lung injury during polymicrobial sepsis.31 32 In trauma patients and mice, ILC2s promote IL-5 expression in neutrophils to exacerbate lung injury.7 The data presented here are the first to demonstrate that patients with HS have a strong trend towards enriched accumulation of ILC2s in the circulation compared with healthy control subjects. Expansion of ILC2s results in type 2 cytokines release and eosinophil infiltration in the lungs, and subsequently aggravated lung injury. ILC2-derived IL-5, IL-9 and IL-13 are responsible for eosinophil infiltration into lung based on ours and others observations.33 This is further evident by our results that replenishing ILC2s in Hmgb1−/− or Rage−/− mice restored eosinophil infiltration in the lungs at 24 hours after HS.
The cytokines IL-33, IL-25 and thymic stromal lymphopoietin (TSLP) released from damaged epithelial cells or activated myeloid cell are important DAMPs for ILC2 expansion.34 35 Extracellular HMGB1 serves as a DAMP to activate immune and inflammatory responses.36 We observed that HMGB1 levels were significantly elevated in patients with HS and mice that were correlated with the expansion of ILC2s. We further demonstrated that rHMGB1 i.t. into WT mice induced ILC2 accumulation in the lungs. Similarly, HS failed to induce ILC2 proliferation in Hmgb1−/− mice or in WT mice treated with anti-HMGB1 antibody. These results establish a novel role for HMGB1 in modulating ILC2 expansion in the lungs following HS.
Receptors for HMGB1 include TLR4, TLR9 or RAGE.12 18 In different pathophysiological conditions, HMGB1 selectively signals by binding to particular receptors through mechanisms that are not yet clear. Some studies suggest that HMGB1-RAGE is instrumental for cell survival, whereas HMGB1-TLRs may be responsible for proinflammatory cytokine production.17 36 In this study, we first showed that RAGE constitutively expressed in ILC2 precursors in BM and mature ILC2s from various lymphoid, mucosal and adipose tissues. We then found a prominent role for RAGE in mediating HMGB1-induced ILC2 proliferation, since the expansion of ILC2s in the lungs was significantly prevented in Rage−/−, but not in Tlr4−/− or Tlr9−/− mice following HS. We also addressed how HMGB1-RAGE affects mitochondrial functional integrity in ILC2s following HS. Compared with the control group, we observed that ILC2s from Hmgb1−/− or Rage−/− mice exhibited decreased mitochondrial mass and mitochondrial depolarisation. We further indicated that Bcl-2, an important antiapoptotic protein, was significantly suppressed in ILC2s in Hmgb1−/− and Rage−/− mice after HS whereas, Bax, a proapoptotic protein, was increased in ILC2s in Hmgb1−/− and Rage−/− mice post-HS. These data suggest that HMGB1-RAGE signalling is required for maintaining mitochondrial homeostasis and promotes ILC2 survival through attenuating mitochondrial mediated apoptosis.
Our study has limitations. First, we were unable to obtain lung tissue samples from patients with HS to directly show elevated ILC2s in the lungs. Second, by using global HMGB1 knockout and RAGE knockout mice, this study is unable to exclude indirect roles for HMGB1 in inducing ILC2 expansion.
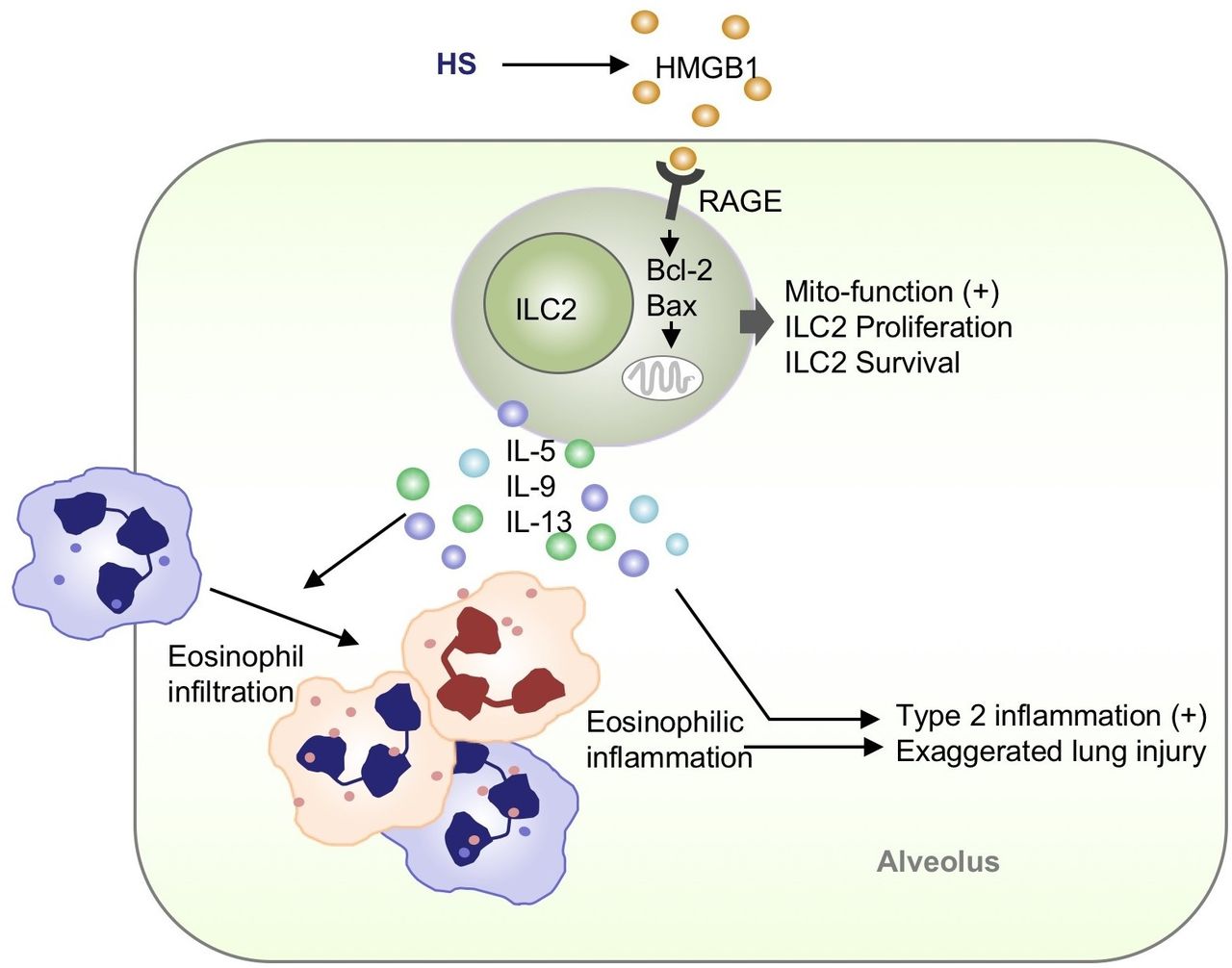
In summary, this study reveals a previously unidentified mechanism by which ILC2s through promoting type 2 immunity aggravate post-HS ALI (figure 9). Targeting HMGB1-RAGE signalling and therefore ILC2 expansion should present an important therapeutic strategy for prevention and treatment of HS-induced ALI.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
High-mobility group box 1 (HMGB1), released during haemorrhagic shock (HS) process, induces lung type 2 innate lymphoid cells (ILC2s) expansion in a receptor for advanced glycation end products (RAGE)-dependent manner. HMGB1-RAGE signalling promotes ILC2 proliferation and decreases ILC2 death through regulating mitochondrial function. Accumulation of ILC2s in the lungs results in initiation of type 2 immune responses, including secretion of interleukin (IL)-5, IL-9 and IL-13, and eosinophil infiltration into the lungs, both of which augments lung inflammation after HS.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
The human study was approved by Research Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (2018-988). The animal protocol was approved by the Institutional Animal Care and Use Committees of the University of Pittsburgh and VA Pittsburgh Healthcare System.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
KZ and YJ contributed equally.
Presented at Partial results of this study were previously presented as an abstract at the 17th International Congress of Immunology, held in China, Beijing. Date 19-23 October 2019. Eur J Immunol 2019;49:432-3. Meeting Abstract number: P0125.
Correction notice In the original version of this article, some H&E images in figure 8, including panel A (Hmgb1-/- sham, and Rage-/- sham) and panel B (control IgY sham, anti-HMGB1 sham, and anti-HMGB1 HS 24h), were presented incorrectly. Figure 8 has been updated with the correct images. All the results have been rescored and reanalysed. The results previously reported as statistically significant remain unchanged. There is no associated change to the text of the article. The authors deeply regret this error.
Contributors KZ and YJ designed and performed the research, and analysed the data; DL, JW, YW, XW and JH performed the research; MS, YL, TB, MW, QS and JF planned the project and conceived the experiments; KZ, YJ, XF and JF conceived the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by the National Institutes of Health Grant R01-HL-079669 (JF and MAW), National Institutes of Health Grant R01-HL-139547 (JF and MAW), National Institutes of Health Grant R01HL076179 (JF), VA Merit Award 1I01BX002729 (JF), VA BLR&D Award 1IK6BX004211 (JF), National Natural Science Foundation of China 81501638 (KZ), 81971809 (YJ), 81720108025 (XF) and National Institutes of Health Grant R01GM102146 (MJS).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.