Article Text

Abstract
Chronic obstructive pulmonary disease (COPD) is a major global health concern with few effective treatments. Widespread destruction of alveolar tissue contributes to impaired gas exchange in severe COPD, and recent radiological evidence suggests that destruction of small airways is a major contributor to increased peripheral airway resistance in disease. This important finding might in part explain the failure of conventional anti-inflammatory treatments to restore lung function even in patients with mild disease. There is a clear need for alternative pharmacological strategies for patients with COPD/emphysema. Proposed regenerative strategies such as cell therapy and tissue engineering are hampered by poor availability of exogenous stem cells, discouraging trial results, and risks and cost associated with surgery. An alternative therapeutic approach is augmentation of lung regeneration and/or repair by biologically active factors, which have potential to be employed on a large scale. In favour of this strategy, the healthy adult lung is known to possess a remarkable endogenous regenerative capacity. Numerous preclinical studies have shown induction of regeneration in animal models of COPD/emphysema. Here, we argue that given the widespread and irreversible nature of COPD, serious consideration of regenerative pharmacology is necessary. However, for this approach to be feasible, a better understanding of the cell-specific molecular control of regeneration, the regenerative potential of the human lung and regenerative competencies of patients with COPD are required.
- lung repair
- regeneration
- regenerative pharmacology
- adult stem cells
- retinoic acid
Statistics from Altmetric.com
The need for a regenerative pharmacological approach in COPD
Chronic obstructive pulmonary disease (COPD) is highly debilitating, often terminal, and widely prevalent. In 2015, it caused 5% of all deaths globally, and the burden of disease is increasing rapidly; currently affecting over 200 million people worldwide, the WHO estimates that COPD will become the third leading cause of death by 2020. According to estimates by the European Respiratory Society, the total annual cost of COPD in Europe is €38.7 billion.
Clinically, COPD is characterised by progressive loss of lung function with airflow obstruction not fully reversible with bronchodilators. These features arise from persistent inflammation associated with bronchitis, small airways disease and alveolar tissue destruction in varying combinations and severities.1 A landmark study linking thoracic CT, and microCT of lung tissue, provided compelling evidence that destruction of small airways is a major contributor to increased peripheral airway resistance in COPD, revealing a significant drop in the number of terminal bronchioles in mild disease (GOLD stage I) with up to 89% reduction in terminal bronchiole number in severe disease (GOLD IV).2 This pathological finding provides an anatomical explanation for the lack of disease-modifying effect of conventional anti-inflammatory treatments in COPD and calls for a dramatic re-evaluation of treatment strategies.
Current approaches focus on prevention of further lung damage. On a population level, tobacco control and environmental air pollution legislation are proven and effective public health interventions.3 Smoking cessation and pulmonary rehabilitation, together with prevention and prompt treatment of infective exacerbations, are individual evidence-based therapies that aim to reduce the rate of lung function decline in patients with COPD.4 Symptomatic treatment, including lung volume reduction, can improve lung mechanics and quality of life in selected patients.
A more ambitious aim is reversal of established lung disease with lung repair or regeneration strategies, which collectively represent a long-term research goal that may provide a novel approach for patients with COPD. To date, clinically tested regenerative strategies include mesenchymal stromal cell (MSC) or tissue engineering-based therapies. MSCs exhibited promising anti-inflammatory and prorepair properties in vitro and in rodent models of emphysema; however, clinical trials using MSCs for COPD are yet to demonstrate clinical efficacy.5 Tissue engineering using autologous airway basal cells has produced tracheas designed for transplantation, although clinical application has met mixed success.6 Bioengineered whole lungs were successfully transplanted into rodents; however, these lungs failed due to incomplete cellularisation and barrier defects leading to fluid exudation.7 According to a recent study in pigs, enhancing vascular integrity in bioengineered lung tissue ex vivo might help improve post-transplantation tissue survival in vivo;8 nonetheless, bioengineering lungs for transplantation in man remains a major challenge largely due to the complex cellularity and architecture of human lung. An alternative and perhaps more tractable approach may be to harness endogenous lung repair or regeneration pathways. Pharmacological manipulation of repair or regeneration has been demonstrated using small molecules in a variety of in vitro and in vivo lung injury models.
Mechanisms of tissue destruction in COPD
Tissue destruction in COPD arises due to a chronic proinflammatory state caused by continual exposure to inhaled pollutants, particularly cigarette smoke. Inflammatory cells are recruited that secrete proteolytic enzymes, causing degradation of elastin and other structural proteins; neutrophils and macrophages additionally create a highly oxidative microenvironment, overwhelming the lung’s endogenous antioxidant mechanisms thereby causing cell and DNA damage.9 These processes may be exacerbated by reduced cellular proliferative capacity, or senescence, with advancing age; persistent senescent cells remain metabolically active and exhibit a senescence-associated secretory phenotype, secreting proinflammatory cytokines.10 Lung function decline in COPD is hastened in subgroups of patients by exacerbations, acute episodes of accelerated deterioration triggered by viral or bacterial respiratory infections. Strategies that reduce the frequency and severity of exacerbations include inhaled steroids and long-acting bronchodilators.11
Only a minority of smokers (~20%) eventually develop COPD, and thus other influences such as early life exposures and genetic factors may increase susceptibility to the effects of smoke in some patients. Genome-wide association studies have identified numerous genetic loci associated with airflow limitation, both in the general population and in patients with COPD; however, the relationship between environmental exposures and genetic predisposition in COPD is complex and incompletely understood.12
Adult lung regeneration: evidence from animal models and feasibility in man
Tissue regeneration can be defined as a process of growth and renewal leading to restoration of tissue structure and function following natural degeneration, injury or disease. The healthy adult mammalian lung possesses an under-recognised capacity for plasticity and regenerative growth. Calorie-restricted adult rodents show reductions in alveolar number and increased alveolar volume due to controlled alveolar septal apoptosis; ad libitum refeeding restored alveolar dimensions to pre-restriction levels.13 Surgical oophorectomy in adult female mice leads to alveolar simplification, which is reversible by oestrogen administration.14 A dramatic example is postpneumonectomy lung growth, in which excision of sufficient volume of lung tissue causes remaining lobes to grow into the resulting cavity, with lung cell proliferation, elastogenesis, angiogenesis and new acinar development leading to lung function recovery and normal alveolar dimensions; this phenomenon has been observed in both rodents and larger mammals.15
The capacity and limits of lung regeneration in humans are yet to be fully elucidated. However, pneumonectomy in children has long been recognised to lead to compensatory growth of the remaining lungs, with concomitant age-dependent improvements in function.16 In an enlightening case study of a young adult female following right pneumonectomy, serial CT imaging over a 15-year follow-up period revealed an expected enlargement of the remaining left lobes but with increased lung tissue density; analysis of acinar-airway dimensions using 3helium MRI suggested the increase in tissue density was consistent with neoalveologenesis rather than enlargement of existing alveoli.17 Adult human lungs may thus possess a greater capability for large-scale cellular and structural reparative remodelling than previously thought.
A recent large, prospective cohort study of intensive care unit patients reported that 60.4% of those who develop acute respiratory distress syndrome (ARDS) survive hospitalisation.18 Remarkably, despite the dramatic parenchymal damage associated with ARDS, pulmonary function in survivors at 5 years is generally normal to near normal.19 Though direct assessment of tissue regeneration in survivor lungs remains technically challenging, lung tissue sections from non-survivors revealed cellular proliferation characteristic of regenerative processes.20 21 Thus, endogenous regenerative mechanisms appear conserved in adult human lung, which may be amenable to pharmacological targeting in disease. Future longitudinal studies using novel, non-invasive high-resolution imaging techniques in patients during resolution of ARDS, or following pneumonectomy, may further define both the capacity and limits of adult human lung regeneration.
Cellular mechanisms of adult lung regeneration
Insights from animal models have revealed stem/progenitor cell populations located throughout the lung capable of responding to injury and repairing damaged tissue.22–24 In mice, the pseudostratified epithelium of the trachea is maintained by basal cells, which function as progenitor cells able to self-renew and generate secretory and ciliated airway cells.25 In the distal mouse lung, club cells and surfactant protein-C (SPC)-expressing alveolar type 2 (AT2) cells can re-enter the cell cycle to maintain airway and alveolar epithelium, respectively, during steady-state homeostasis and following mild injury.26 27 Evidence from mouse models suggests distal lung regeneration following severe injury, such as sublethal influenza infection, may be performed by AT2 cells28 and/or by distal multipotent quiescent stem cells which can regenerate both airway and alveolar epithelium.29 30 Emerging evidence reveals a remarkable phenotypic plasticity of adult lung epithelial cells.31 Selective ablation of basal cells in the adult mouse trachea induced club cells to dedifferentiate to replenish the basal cell pool.32 HOPX+ alveolar type 1 cells dedifferentiated into AT2 cells in adult mice in vivo during postpneumonectomy lung regeneration,33 and club cells isolated from adult mouse airways differentiated into AT2 cells when cultured with an alveolar mesenchymal population.34
In adult human lung, basal cells, distributed throughout the conducting airways extending to the terminal bronchioles, function as progenitor cells for maintenance and repair of airway epithelium.35 Basal cells of the proximal and distal human lung share similar morphology, yet in vitro experiments have revealed differences in differentiation capability; for example, distal basal cells appear to have alveolar lineage potential.29 Moreover, SPC+ progenitor cells with alveolar lineage potential have been isolated from human distal lung.26 28
Studies of postpneumonectomy lung regeneration in mice revealed widespread proliferation of mesenchymal cells, including endothelial cells and fibroblasts;36 however, mesenchymal lung stem/progenitor cells are poorly characterised. Recently, a mesenchymal population expressing Lgr6 (a Wnt signalling co-receptor), and a possibly overlapping population expressing the Wnt target gene Axin2, were both shown to regenerate airway smooth muscle in adult mice.34 37 C-kit expression characterised endothelial progenitor cells resident in adult mouse lung that contributed to microvascular maintenance and repair following pneumonectomy.38 A stromal population termed lung-resident mesenchymal stem cells (LR-MSCs) proliferated following pneumonectomy in adult mouse lung,39 and a possibly analogous LR-MSC population isolated from human peripheral lung were enriched in clonogenicity and multi-lineage potential.40
The precise progenitor populations responsible for adult human lung maintenance and repair, and the extent to which these populations repair different types of injury, is a matter of intense investigation. In principle, endogenous stem/progenitor cells could be pharmacologically stimulated to repair or regenerate diseased lung tissue, although caveats include the challenge of how to target them specifically; their presence or functionality in lungs of patients with COPD remains to be fully characterised.
Molecular control of adult lung regeneration
Proliferation, differentiation and reorganisation of stem/progenitor cells during adult regeneration may in part recapitulate developmental processes.41 Signalling pathways that control lung development are often reactivated in adult lung regeneration, including Wnt/β-catenin, notch, retinoic acid (RA), bone morphogenic protein, fibroblast growth factors (FGFs) and hedgehog signalling pathways.22–24 Mouse studies have implicated a network of promitogenic pathways that control adult lung stem/progenitor proliferation, including FGF receptor 2 (FGFR2) and epidermal growth factor receptor that signal via mitogen activated kinase (MAPK) and phosphatidylinositol 3-kinase-protein kinase B.42–44 Mechanisms also actively limit lung cell growth, for example, FGFR1 limited basal cell proliferation in adult mouse trachea via SPRY2-mediated inhibition of MAPK,45 and sonic hedgehog secreted by lung epithelial cells maintained quiescence of underlying mesenchyme in adult mouse lung.46
Recent single-cell RNA sequencing of adult lung tissue revealed conservation of an injury-responsive hypoxia/notch/keratin-5 programme in epithelial progenitors from human and mouse lung,47 and thus local lung hypoxia during infection may trigger regeneration by specific lung-resident stem cells. Whether these programmes are conserved in COPD, in which microvascular defects may lead to local lung hypoxia, remains unknown. Interestingly, sensitivity to Wnt and FGF pathway stimulation in vitro was conserved in SPC+ alveolar epithelial progenitor cells from both human and mouse lung.28 Thus, while most data about lung regenerative mechanisms derive from mouse studies, the limited human data available are consistent with conservation of regenerative mechanisms in adult human lung tissue, supporting the feasibility of targeting these pathways to induce regeneration in disease. Additional human studies are required to clarify the degree of any overlap between animal and human lung.
To what extent is regenerative capability maintained in the COPD lung?
Disturbed regenerative capacity in COPD lung
The local microenvironment in COPD lung tissue is proinflammatory and pro-oxidative due to increased generation of reactive oxygen species (ROS), which could interfere with molecular pathways required for stem cell activation. ROS induce cellular defense mechanisms by upregulating free radical scavenging enzymes (eg, manganese superoxide), and DNA damage repair genes (eg, Gadd45) in part via activation of the transcription factor forkhead box O (FOXO). In vitro, ROS diverted β-catenin away from T cell factor mediated transcription towards FOXO, thus inhibiting the β-catenin transcriptional programme.48 Thus, increased ROS production could impair Wnt/β-catenin dependent repair in the COPD lung. Accordingly, a decrease in nuclear β-catenin, suggesting reduced Wnt signalling activity, was identified in AT2 cells in COPD lung.49 Thus, pharmacotherapies aimed at stimulating endogenous repair pathways may additionally need to address a potentially hostile local microenvironment.
While stem/progenitor cell populations may survive in COPD lung, they might acquire defects in disease, compromising their function. For example, telomere shortening was described in smokers with and without COPD and AT2 cell senescence increased in patients with emphysema.50 51 Smoke increased basal cell propensity for squamous metaplasia and epithelial-mesenchymal transition and caused loss of junctional barrier integrity via EGF induction in neighbouring cells.52 Basal cells maintain human airway epithelium through clonal expansion; in smokers, basal expansion was accelerated leading to more rapid clonal consolidation, which could generate premalignant field defects with aberrant regenerative ability.53 Moreover, smoke exposure led to loss of regional identity and reprogramming of distal airway epithelial cells to more proximal characteristics.54 Thus, constant insults may exhaust the capacity of stem cells to respond to injury, thereby contributing to airway epithelial defects in COPD.55
Growing evidence implicates dysregulation of pathways that control early lung development in the pathogenesis of COPD. For example, gene expression profiling revealed reduced activity of both Notch and Wnt signalling pathways in airway epithelial cells of non-diseased smokers and smokers with COPD.56 57 Although these pathways may be amenable to pharmacological targeting, molecular changes in specific cell populations in the COPD lung may desensitise them to proregenerative signals. Lung fibroblasts isolated from patients with COPD exhibited reduced gene expression for cellular RA binding protein-1, required for intracellular RA transport, and synthesised less elastin in response to RA compared with non-diseased fibroblasts from cancer resections.58 In emphysematous lung tissue, we found increased gene and protein levels of cytochrome P450 subfamily 26 A1 (CYP26A1), an endothelial-expressed enzyme which selectively degrades RA; increased enzyme activity would be expected to decrease endothelial RA signalling activity and contribute to failure of regeneration.59
Apoptosis of AT2 cells and microvascular endothelial cells was increased in lungs of patients with COPD/emphysema, suggesting that crucial lung stem/progenitor populations could be depleted in severe disease.60 Decreased basal cell numbers and a propensity for goblet differentiation in surviving basal cells was reported in airway biopsies from patients with COPD compared with patients without COPD,61 and bronchial epithelia of smokers contained fewer club cells compared with non-smokers.62 Mechanisms of small airway destruction in human COPD are poorly understood,2 35 and whether cells that can regenerate small airways survive in COPD is currently unclear. A recent report of transcriptional and functional characterisation of human small airway epithelium may help elucidate this issue;63 similar studies in COPD lung could be informative. The cell types that comprise the simple cuboidal epithelium of the respiratory bronchioles in the adult human lung have yet to be fully characterised. In the most distal regions of the emphysematous lung, widespread alveolar septal wall destruction could deplete matrix substrate required for regeneration.
Studying adult lung regeneration in vivo in the context of COPD
Animal models of COPD typically involve inhalation or instillation of agents into the lung.64 Most faithful to human COPD is chronic exposure to cigarette smoke for several months, usually in rodents or guinea pigs, which induces pathology that resembles mild human COPD (GOLD stage I or II) including emphysema, airflow obstruction and airway remodelling. Intratracheal elastase is widely used to model emphysema in rodents and causes acute haemorrhage, inflammation and proteolysis of alveolar septal elastin leading to persistent airspace enlargement. Other models include repetitive intratracheal lipopolysaccharide (LPS) or lung-specific inducible manipulation of specific genes.64 Alternatively, halting alveolar development, for example, with perinatal administration of dexamethasone, causes enlarged alveolar spaces that persist into adulthood, resembling adult bronchopulmonary dysplasia (BPD), without ongoing destruction.65 Major limitations of animal COPD models relate to important anatomical differences with human lung. Crucially, mice lack respiratory bronchioles; the smallest airways are large relative to the size of the animal.66 Respiratory bronchioles, seen in generations 15–23 in humans, are major sites of obstruction in COPD,67 and thus while animal COPD models can mimic emphysema, small airway disease is poorly represented.64 Furthermore, pathology of severe disease is challenging to recapitulate, and while disease progresses after smoking cessation in humans, smoke withdrawal in animals halts pathological progression.64 Despite limitations, numerous studies have demonstrated the promising ability of pharmacological agents to reverse pathological features in these models.
Regenerative pharmacology in COPD
In pioneering experiments, Massaro and Massaro demonstrated that in an adult rat model of elastase-induced emphysema, administration of all-trans RA (ATRA) stimulated alveolar regeneration, measured by more numerous, smaller alveoli and improved elastic recoil.68 ATRA, derived from dietary vitamin A, is the endogenous agonist for RA receptors (RARs), type 2 nuclear receptors which heterodimerise with retinoid X receptors (RXRs), through which ATRA regulates expression of genes involved in cell growth and differentiation. RA induced alveolar regeneration in adult rat and mouse emphysema and BPD models in several independent studies, improving alveolar dimensions and some lung function parameters;65 69–71 however, other studies failed to find an effect of RA in elastase or cigarette-smoke models, possibly due to different sensitivities to RA between animal strains.72 73
In adult rats, keratinocyte growth factor (KGF; FGF7) administered after pneumonectomy augmented alveolarisation,74 and in adult mice with elastase-induced emphysema, palifermin (a truncated version of KGF) stimulated alveolar regeneration, improving alveolar dimensions and relieving airflow limitation.75 In a human model of ARDS with inhaled low-dose LPS, intravenous palifermin increased alveolar surfactant protein D and matrix metalloproteinase-9, suggestive of AT2 cell proliferation, and altered matrix repair programmes, respectively.76 KGF signalling appears intact in human emphysema, as KGF ligand and receptor protein were unchanged in lung tissue of patients with emphysema compared with normal patients and healthy smokers,77 supporting the potential of KGF signalling as a therapeutic target for COPD.
Wnt/β-catenin signalling regulates progenitor cell renewal in numerous adult mammalian organs; in adult mouse lung, Wnt ligand-mediated autocrine signalling promoted AT2 proliferation following injury.78 Pharmacological Wnt/β-catenin activation reversed alveolar architectural defects in a mouse elastase model.49 Expression of the Wnt receptor Frizzled 4 (FZD4) was decreased in COPD lung tissue homogenates and in AT2 cells derived from patients with COPD,79 suggesting that prospective therapeutics targeting the Wnt/β-catenin pathway may need to act downstream of Wnt receptors. Potentially useful examples include pharmacological inhibitors of the cytosolic kinase GSK3, which cause β-catenin accumulation and thus activation of the Wnt/β-catenin pathway.80
Hepatocyte growth factor (HGF) regulated adult alveolar maintenance in mice.81 Administration of HGF stimulated alveolar regeneration in elastase-treated rats, enhancing lung vascularisation and improving exercise tolerance and gas exchange.82 Fibroblasts from human emphysematous lung produced less HGF than control lung fibroblasts,83 suggesting potential for therapeutic normalisation of lung tissue HGF levels in human chronic lung disease.84
Adrenomedullin has pleiotropic roles including vasodilation, angiogenesis and endothelial cell growth and survival. In neonatal mice, administration of adrenomedullin protected against the effects of hyperoxia, preserving lung capillary density and alveolar development,85 while in adult mice with elastase-induced emphysema, adrenomedullin improved alveolar dimensions, lung volume and static compliance.86 Adrenomedullin stimulated repair of human bronchial epithelial cells in vitro, supporting its relevance to human lung physiology.87
Basic fibroblast growth factor (bFGF; FGF2) is a proangiogenic, heparin-binding FGF family member. In adult mice, subcutaneous bFGF injections preserved alveolar dimensions after repeated LPS administration.88 Intratracheal administration of bFGF-soaked microspheres to adult rats and dogs with elastase-induced emphysema improved alveolar dimensions, lung microvessel density and arterial oxygenation.89
Vascular endothelial growth factor (VEGF) is a proangiogenic, endothelial-specific mitogen. VEGF administration enhanced postpneumonectomy alveolar growth in mice.90 In rats with enlarged alveoli due to neonatal hyperoxia, VEGF administered in adulthood stimulated alveolar regeneration,91 and overexpression of VEGF promoted repair and increased lung capillary density.92
The cellular mechanisms of regeneration in these studies are unclear, but notably, the agents used can target different lung cell types. Studies by us and others suggest an unexpected complexity to the cellular landscape of RA responsiveness in the lung: in vitro, RA stimulated elastin synthesis in lung fibroblasts93 and angiogenesis in lung microvascular endothelial cells,59 whereas RA restricted adult lung epithelial progenitor cell proliferation while promoting differentiation in a mouse lung organoid model.94 Moreover, VEGF was mitogenic for both endothelial and alveolar epithelial cells,95 whereas KGF is an epithelial-specific growth factor;96 thus, there may be multiple cellular routes to inducing regeneration in adult lung. However, whether stimulating one population is sufficient to coordinate growth of multiple cell types requires further investigation (figure 1). This might be important for the COPD lung, which may lack specific cell types depending on disease phenotype. Given the recent findings that external cues may instruct phenotypic plasticity of adult lung epithelial cells,31 an intriguing possibility is that depleted progenitor populations in COPD could be restored by pharmacologically reprogramming surviving epithelial cells into the required lineages.
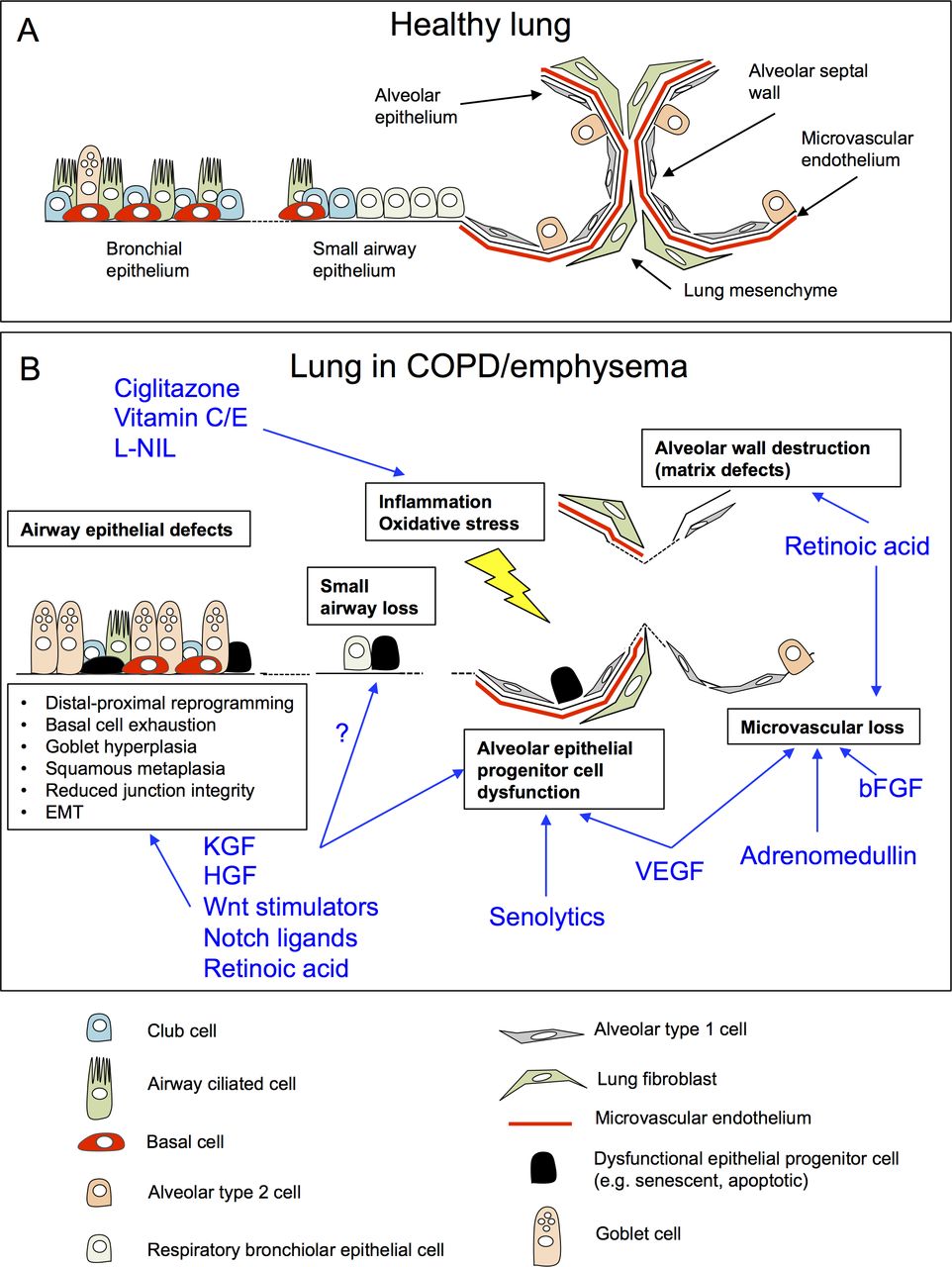
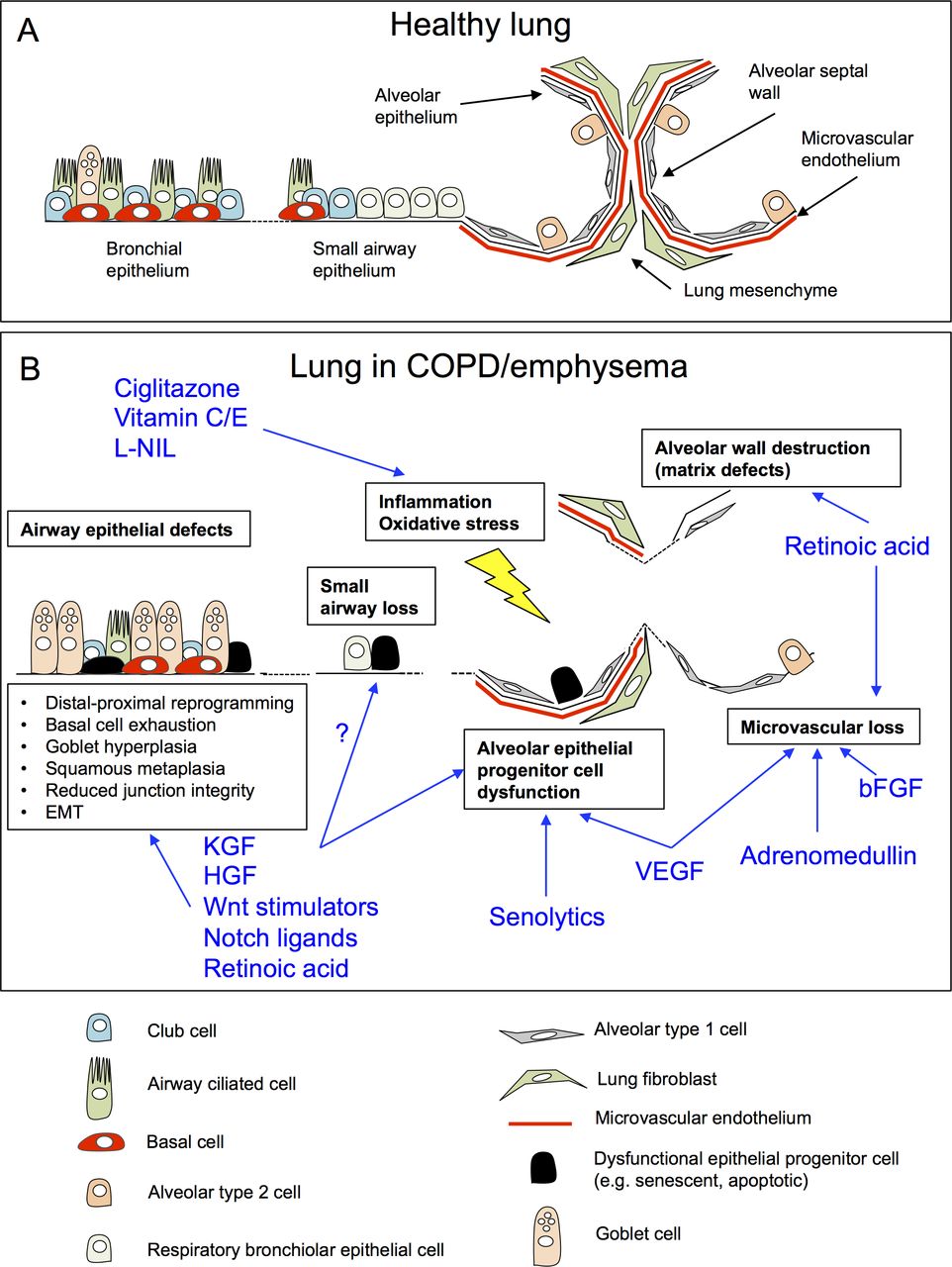{kind=link}
Challenges and opportunities for regenerative pharmacology in COPD/emphysema. (A) Schematic of the cellular architecture of healthy adult lung tissue. (B) Cellular and architectural defects in the lung in COPD/emphysema (boxes), and putative mechanisms of action by proregenerative agents identified from pre-clinical animal models of COPD/emphysema (blue; references within text). bFGF, basic fibroblast growth factor; COPD, chronic obstructive pulmonary disease; HGF, hepatocyte growth factor; L-NIL, N6-(1-iminoethyl)-L-lysine; KGF, keratinocyte growth factor; VEGF, vascular endothelial growth factor.
In mice exposed to cigarette smoke for 5 months, combining the last 2 months with intranasal ciglitazone, an anti-inflammatory peroxisome proliferator-activated receptor-γ agonist, restored alveolar dimensions and lung volume compared with controls.97 Similarly, in a cigarette-smoke mouse model, treatment for 2 months after smoke cessation with a combination of the antioxidants vitamin C and E restored alveolar dimensions,98 and in mice exposed to cigarette smoke for 8 months, causing pulmonary hypertension and emphysema, administration of N6-(1-iminoethyl)-L-lysine, an inhibitor of inducible nitric oxide synthase for an additional 3 months completely restored alveolar number and regenerated septal elastin fibres.99 These studies are intriguing as they suggest ameliorating a hostile microenvironment with anti-inflammatory, antioxidant or antinitrosative treatments might allow endogenous regenerative mechanisms to restore architectural defects (figure 1).
Clinical trials with regenerative therapies
Therapeutic attempts to stimulate lung regeneration in COPD are limited to four relatively short-term clinical trials with retinoids. Two studies in patients with emphysema used a crossover design with orally administered ATRA.100 101 In both studies, ATRA was well tolerated but no differences were observed in CT, lung function or quality of life scores between treatment groups. Average plasma ATRA levels following drug administration declined over the study period, attributed to autoinduced catabolism,101 so it is unclear whether ATRA reached cell types necessary for repair.
Two parallel-group, placebo controlled trials were conducted with the RAR-γ selective agonist palovarotene. One study used patients with emphysema secondary to alpha-1-antitrypsin deficiency; no differences in CT scores or lung function were observed, although palovarotene protected against the effect of exacerbations on decline of lung density, a measure of emphysema progression.102 The second involved patients with CT-confirmed smoking-related emphysema.103 No significant efficacy was reported overall, although posthoc subgroup analysis found that patients with lower zone disease had reductions in decline of FEV1, gas diffusing and exercise capacity compared with placebo.
While the overall failure of the retinoid trials is disappointing, the findings that an RAR-γ agonist may reduce the rate of decline in a subset of patients, particularly during exacerbations,102 may warrant further investigation. Although these findings are yet to be corroborated in independent studies, they suggest that biological activity of retinoids in patients with emphysema and may reflect regenerative potential. A regenerative pharmacology approach with potential to slow lung function decline may be an acceptable therapeutic intervention. Notably, our recent finding that RA pathway activation restricted adult lung epithelial progenitor cell proliferation while promoting differentiation, highlights the possibility that RA signalling might need to be carefully, transiently repressed, then reactivated in the lung epithelium, to allow regenerative growth and correct differentiation in diseased adult lung tissue.94
Future challenges and therapeutic potential of regenerative pharmacology
The anatomical and physiological differences between mouse and human lung highlight the need for predictive preclinical human models of COPD, which are currently scarce. The extent to which regenerative mechanisms remain competent in human COPD lung remains to be fully characterised. Novel approaches including transcriptional profiling of human lung tissue at single-cell resolution are transforming our understanding of the cellular composition of the adult human lung, an example being the recent discovery of the pulmonary ionocyte;104 105 such an approach applied to COPD lung may provide new insights into regenerative competence of diseased lung tissue.106 Air-liquid-interface or lung-on-chip methods that recapitulate human small airways may help to elucidate mechanisms to regenerate terminal bronchioles in COPD.63 107
Given the complex nature of endogenous lung repair, it is likely that activation of multiple signalling pathways is required to stimulate regeneration in COPD; a better understanding of cell-specific roles of molecular pathways in lung regeneration is needed. The optimal timing, duration and method of delivery of regenerative factors remain entirely unknown. Targeting correct cell populations, specifically and precisely, with the correct pharmacological tools will be an additional challenge. For example, sequential activation of stem cell division, then differentiation, in concert with induction of angiogenesis, may be needed. As dysregulated proteolysis may deplete matrix substrate in severe COPD, a successful regenerative therapy is likely to require controlled synthesis and organisation of matrix to provide regenerating cells an endogenous scaffold for restoration of functional tissue.
As adult regenerative processes in part reiterate developmental mechanisms, a successful regenerative intervention may need to orchestrate a programme of changes that recapitulates lung formation. Coordinating proportionate growth of multiple cell lineages will help to mitigate aberrant tissue remodelling which could further impair lung function, whereas focused and controlled induction of differentiation will be critical to avoid cancer associated with activation of mitogenic pathways. Importantly, key molecular pathways must be activated and deactivated appropriately during regeneration; aberrant activation of developmental pathways is implicated in the pathogenesis of chronic lung diseases including IPF.108 Development of therapeutics with well-defined clearance or degradation pathways, and thorough characterisation of in vivo pharmacokinetics in human subjects, will aid in fine tuning key signalling pathways for therapeutic applications.
Of note, it may be possible to use pharmacological strategies to augment endogenous tissue repair without inducing regenerative cellular growth. For example, in fibrotic lung tissue slices generated from adult mice, senolytic drugs, which specifically target senescent cells, decreased senescence and fibrotic markers and increased epithelial markers.109 Expression of the planar cell polarity pathway members Van Gogh-Like 2 (VANGL2) and Scribble were decreased in lung tissue from patients with COPD, and VANGL2 controlled actin microfilament organisation and migration of adult lung epithelial cells,110 suggesting that targeting mechanisms regulating cytoskeletal dynamics could enhance cell migration and wound healing in diseased lung independent of growth.
Personalised regenerative pharmacology
The lowering cost and increasing availability of high-throughput methods may allow personalisation of a regenerative treatment. Endobronchial biopsies from patients with COPD could be subjected to transcriptomic, proteomic or metabolomic profiling to identify the specific molecular phenotype of that patient and provide crucial information about integrity of potential targetable molecular pathways. Single cell transcriptional sequencing could be applied to lung biopsies to reveal progenitor cell populations to be targeted by regenerative therapies. Alternatively, lung organoids could be generated either from patient-derived induced pluripotent stem cells nurtured towards lung cell fate or by isolating stem cells from airway biopsies or nasal brushings.111 This could provide a platform for high throughput compound screens using small compound or biological molecule libraries to expedite identification of personalised regenerative therapeutics.
Conclusion and perspective
The demonstration of lung regeneration in animal models of COPD/emphysema, together with our improving understanding of the cell and molecular biology of regeneration, give hope that pharmacological induction of lung regeneration in man may be possible. Recent studies revealing basal cell defects in COPD have helped improve our understanding of regenerative competence in disease. Additional investigation into their molecular control, and further characterisation of progenitor populations in adult human lung, may reveal novel targets for regenerative therapies. Development of human models of small airways disease, in combination with high-throughput screens with small compound or biological molecule libraries, will aid identification of novel therapeutics with regenerative potential and will hopefully reveal needed insights into mechanisms of small airway regeneration. As the regenerative competence of the COPD lung is likely to depend on disease severity, biomarkers that allow early identification of those patients that will go on to develop progressive lung disease are required to allow optimal opportunity for intervention.
Acknowledgments
We apologise for the many important contributions to regenerative COPD research that we could not include due to space constraints.
References
Footnotes
Contributors All authors: conception and design, manuscript writing, critical reading and revision, final approval of manuscript.
Funding This work was supported by the Lung Foundation Netherlands (Longfonds) under grant no. 6.1.14.009 and by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.