Article Text

Abstract
Background While the international spread of multidrug-resistant (MDR) Mycobacterium tuberculosis strains is an acknowledged public health threat, a broad and more comprehensive examination of the global spread of MDR-tuberculosis (TB) using whole-genome sequencing has not yet been performed.
Methods In a global dataset of 5310 M. tuberculosis whole-genome sequences isolated from five continents, we performed a phylogenetic analysis to identify and characterise clades of MDR-TB with respect to geographic dispersion.
Results Extensive international dissemination of MDR-TB was observed, with identification of 32 migrant MDR-TB clades with descendants isolated in 17 unique countries. Relatively recent movement of strains from both Beijing and non-Beijing lineages indicated successful global spread of varied genetic backgrounds. Migrant MDR-TB clade members shared relatively recent common ancestry, with a median estimate of divergence of 13–27 years. Migrant extensively drug-resistant (XDR)-TB clades were not observed, although development of XDR-TB within migratory MDR-TB clades was common.
Conclusions Application of genomic techniques to investigate global MDR migration patterns revealed extensive global spread of MDR clades between countries of varying TB burden. Further expansion of genomic studies to incorporate isolates from diverse global settings into a single analysis, as well as data sharing platforms that facilitate genomic data sharing across country lines, may allow for future epidemiological analyses to monitor for international transmission of MDR-TB. In addition, efforts to perform routine whole-genome sequencing on all newly identified M. tuberculosis, like in England, will serve to better our understanding of the transmission dynamics of MDR-TB globally.
- tuberculosis
- clinical epidemiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Does combining distinct datasets of Mycobacterium tuberculosis whole genome sequences from around the globe improve understanding of the international spread of multidrug-resistant tuberculosis?
What is the bottom line?
In a global genomic analysis, extensive international spread of multidrug-resistant tuberculosis was observed, including relatively recent movement of strains from varied genetic backgrounds between high-burden and low-incidence countries.
Why read on?
In nearly all examples of multidrug-resistant (MDR) clades that had dispersed internationally, strains from different countries were isolated and sequenced as part of separate investigations, highlighting the power of combining data across diverse studies and the need for international cooperation in order to contextualise the global MDR-tuberculosis epidemic and halt the spread of drug-resistant strains.
Introduction
Multidrug-resistant (MDR) tuberculosis (MDR-TB) is defined when Mycobacterium tuberculosis isolated from a patient exhibit in vitro resistance to both isoniazid and rifampicin, two key first-line antitubercular drugs. With an estimated 480 000 new cases and approximately 250 000 deaths from MDR-TB globally in 2015,1 MDR-TB is a major threat to global TB control efforts. In 2015, the WHO classified 30 countries as high burden for MDR-TB in an effort to better focus attention on the MDR-TB crisis; these countries account for 89% of MDR-TB cases worldwide.2
The global epidemic of MDR-TB is due to both de novo acquisition of resistance during treatment and person-to-person transmission. Globalisation has facilitated movement of people, and consequently movement of drug-resistant strains around the world. Prior investigations have documented transmission of MDR-TB within countries;3–7 however, international dissemination of MDR-TB strains has not been well studied. Few studies in low-burden settings have investigated whether or not MDR-TB was ‘imported’, meaning that it was acquired before immigration to a low-burden country.8 9 A survey of variable number tandem repeat (patterns) molecular fingerprints by the European Centre for Disease Prevention and Control concluded that international transmission of MDR-resistant and extensively drug-resistant (XDR)-TB plays an important role in TB incidence in Europe,10 especially import of Beijing isolates from Eastern European countries.10 11 Other studies have linked MDR-TB and XDR-TB isolates from the U.K. to strains circulating in Russia, Eastern Europe or South Africa5 7 9 12 as well as linking MDR-TB strains in Thailand and the USA.13
Several of these existing studies have looked for international links in a targeted manner.11–13 To date, more comprehensive examinations of the global spread of MDR-TB using whole-genome sequencing have not been performed. In Manson et al (2017),14 we constructed a global whole genome dataset of 5310 diverse M. tuberculosis isolates from patients in 48 countries on five continents. Comparative analysis of these geographically distinct isolates supported the existence and emergence of MDR-TB and XDR-TB throughout the globe and provided a unique opportunity to investigate evolutionary relationships among drug-resistant strains on a global scale. Here, we identify migratory clades of MDR-TB, where MDR strains have disseminated globally and estimate the timing of their geographic divergence.
Methods
We used the global dataset of 5310 strains from Manson et al (2017), together with their database of drug resistance mutations and parsimony analysis to determine phylogenetic nodes at which genotypic drug resistance evolved.14 As described previously, sequence reads were mapped onto M. tuberculosis reference strain H37Rv (GenBank accession number CP003248.2) using BWA V.0.7.1015 and variant calls were performed using Pilon V.1.11.16 For phylogenetic reconstruction, all sites with unambiguous single nucleotide polymorphisms (SNPs) in at least one strain were combined into a concatenated alignment containing 231 898 polymorphic sites. Ambiguous positions were treated as missing data. The concatenated alignment was then used to generate a midpoint rooted phylogenetic tree using FastTree.17 Genotypic MDR was defined by identification of resistance mutations to both isoniazid and rifampicin (online supplementary appendix table 1). Diversity among strains was assessed using pairwise SNP differences (online supplementary appendix figure 1).
Supplemental material
Supplemental material
Clades of MDR-TB were defined as groups of isolates sharing a single shared ancestral evolution of MDR-level resistance that resulted in at least two descendant strains with genotypic MDR. We excluded 8 of 40 (20%) MDR clades with descendants from multiple countries due to their having low bootstrap support that fell below our threshold for inclusion of 0.8. For each high confidence node (bootstrap value ≥0.8) at which genotypic MDR evolved within our global phylogenetic tree, we examined the geographic origins of the descendent strains with respect to country of origin and UN Geoscheme designation.18 Countries were classified as high burden for MDR-TB if they were designated as such by the WHO in the 2016 Global Tuberculosis Report.1
As done previously,19 each strain’s ‘digital’ spoligotype was predicted by determining the presence or absence of 43 unique spacer sequences among the sequence read data.
We defined ‘migrant’ MDR clades as MDR clades in which descendant strains were identified in two or more countries. Migrant clades were further characterised with respect to number of descendants as well as characteristics of descendant strains including spoligotype, country and UN geographic region of isolation, genotypic XDR status and study source. Within migrant MDR clades, we determined the number of SNPs that separated the two most closely related isolates from different countries as a proxy for when the isolates diverged geographically. We estimated divergence times using estimated mutation rates of 0.3 and 0.6 SNPs/genome/year.19
Results
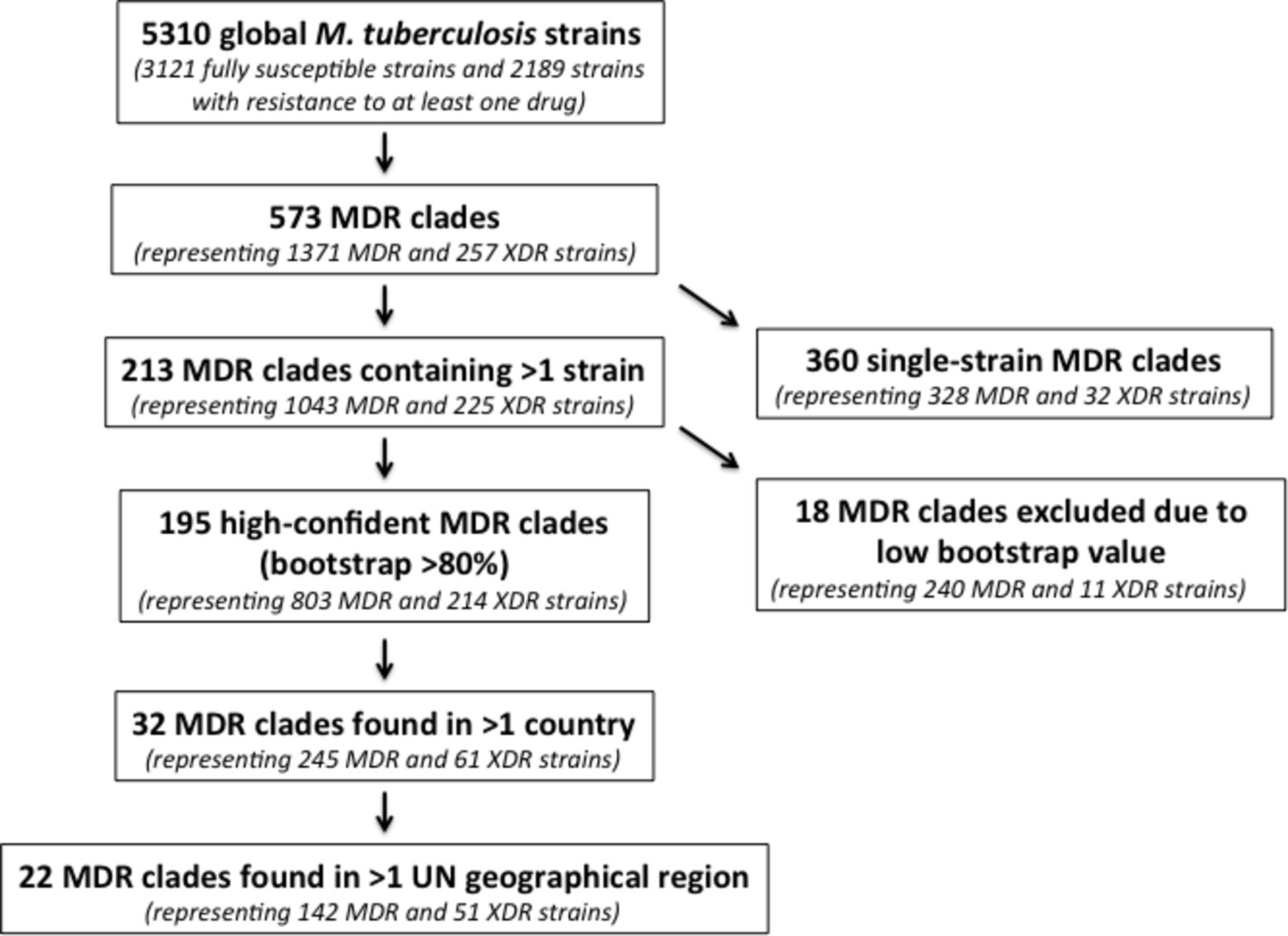
In an analysis of 5310 clinical M. tuberculosis isolates obtained from patients in 48 countries, we identified 573 independent evolutions of MDR-TB.14 Of these 573 independent evolutions of MDR-TB, 18 were excluded from further analysis due to our inability to confidently evaluate phylogenetic structure within that part of the tree (Methods), and 360 were associated with a single MDR isolate. This left us with 195 MDR clades with high-confidence nodes throughout the phylogeny (represented by a total of 1017 member strains) (figure 1), suggesting that MDR-TB had been transmitted between at least two individuals hundreds of times.
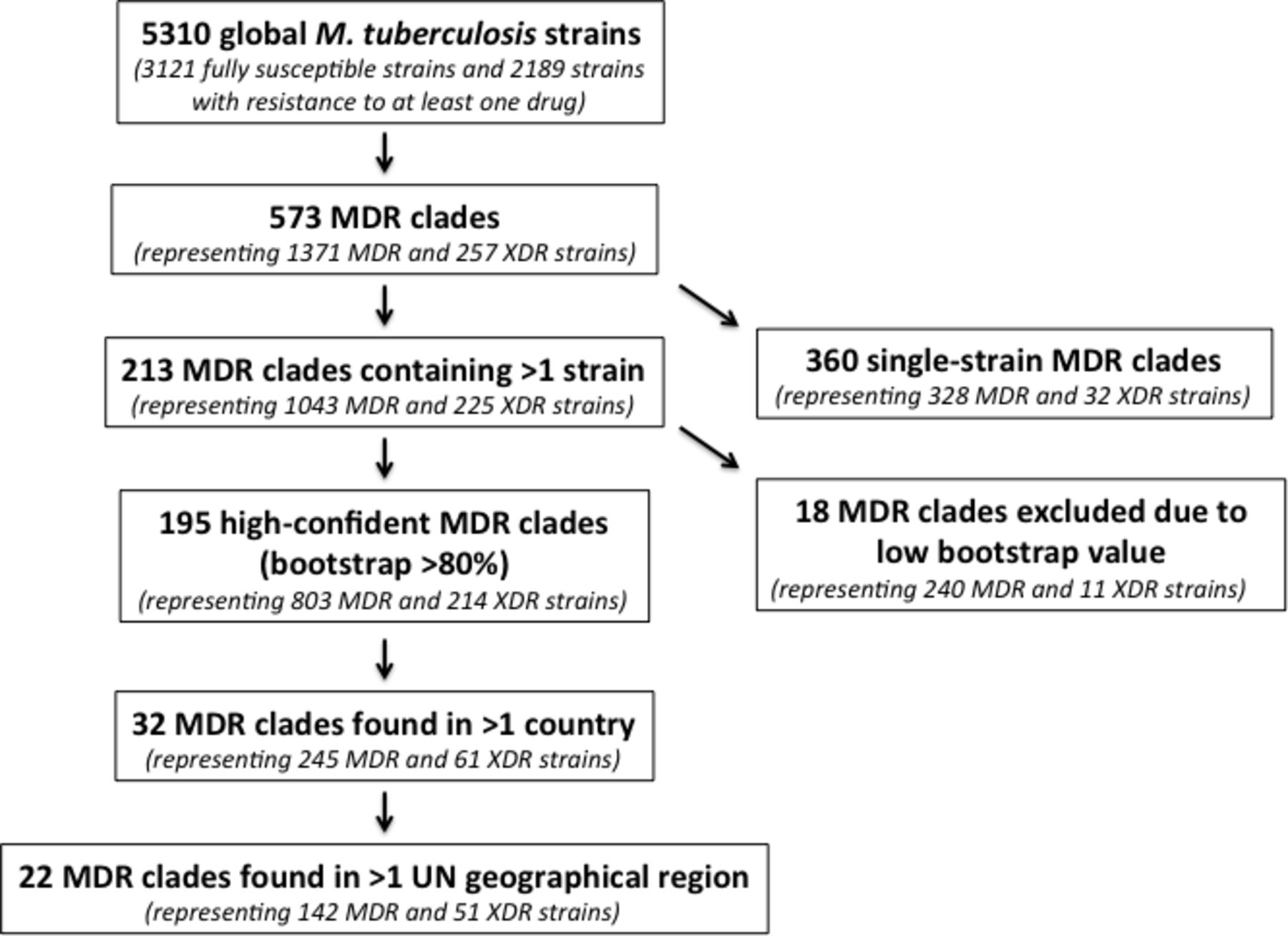
Flow diagram of included study strains, numbers of MDR evolutions, numbers of MDR clades and MDR migrant clades. MDR, multidrug-resistant; XDR, extensively drug-resistant.
We observed descendants of MDR evolution in 24 countries. Seventeen of these countries, in Africa, Asia and Europe, harboured descendants of MDR clades that were isolated in two or more countries, which we define as globally ‘migrant’ MDR clades. Sixteen per cent (32 of 195) of MDR clades, containing a total of 306 isolates, were classified as migrant, indicating repeated global spread of MDR-TB (figure 2; table 1A and B); the remaining 163 MDR clades (84%) were confined to a single country (online supplementary appendix table 2). While we did not observe any examples of internationally migrant XDR clades in this dataset (online supplementary appendix figure 2), de novo emergence of XDR was frequently observed within migrant MDR clades. More than 50% (17 of 32) of migrant MDR-TB clades contained at least one XDR strain. In total, 61 XDR strains were identified within migrant MDR clades.
Supplemental material
Supplemental material
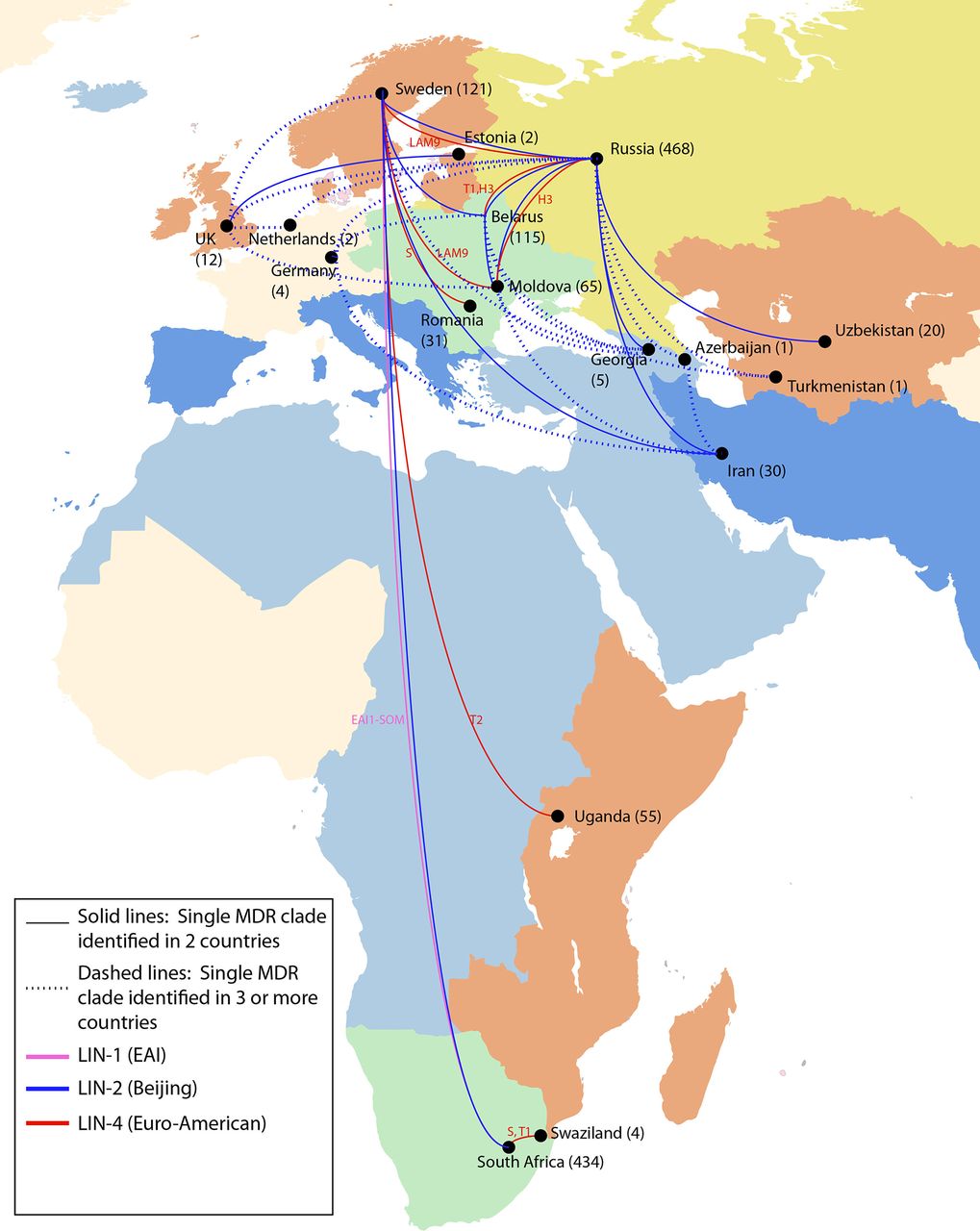
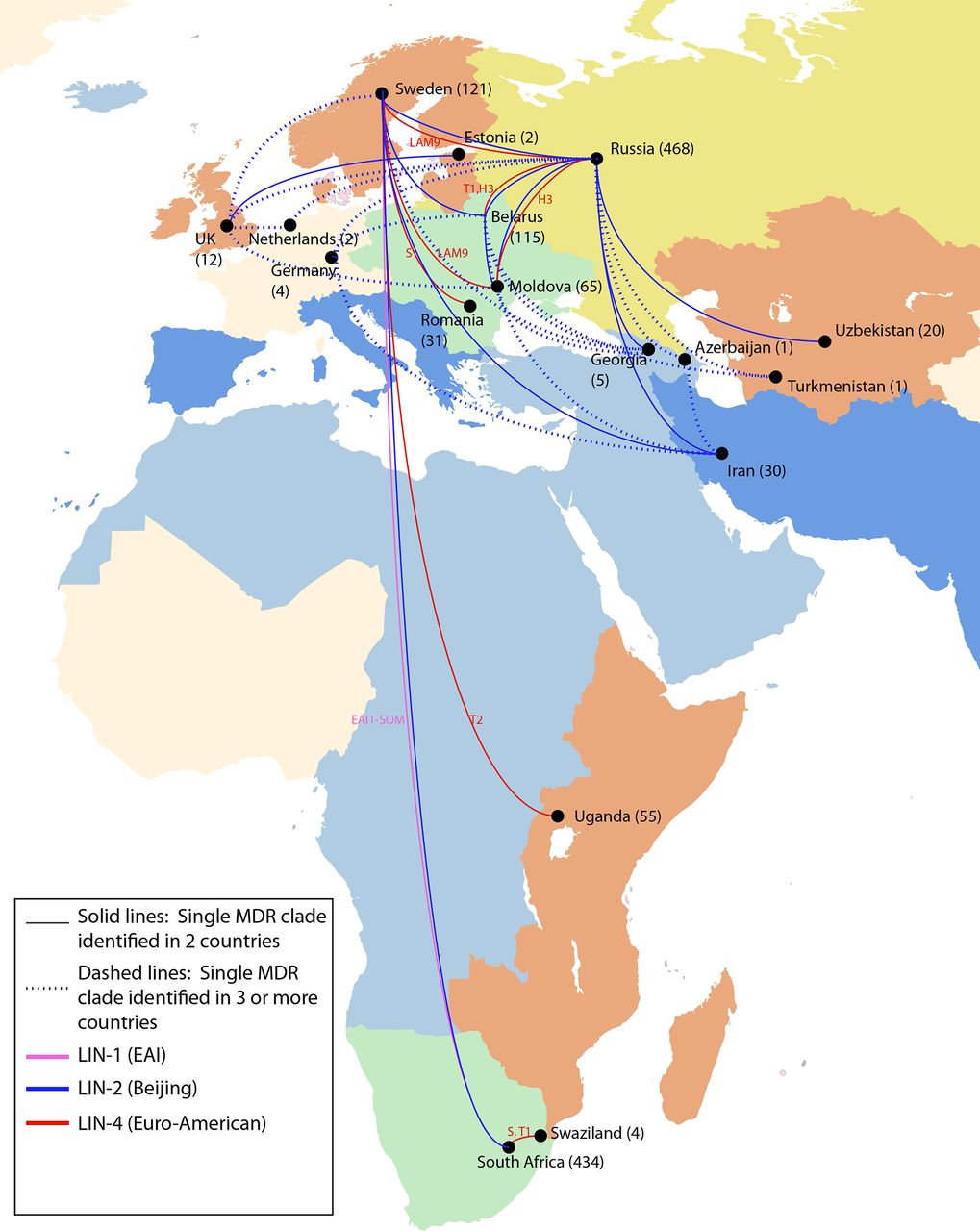
International spread of MDR Mycobacterium tuberculosis strains. In this dataset of 5310 strains, there were 32 examples in which a single evolution of MDR resulted in descendent strains isolated in more than one country, implying movement of MDR strains between countries. The numbers in parentheses indicate the total number of MDR strains in our dataset from each country, of which a subset was involved in geographic movement. The lines showing international spread are coloured by lineage. The spoligotype designation for all lineage 2 strains is Beijing; lineage 1 and 4 spoligotypes are indicated. The map is coloured by UN geographic subregion. MDR, multidrug-resistant.
Movement of MDR strains around the globe
Nearly 70% (22 of 32) of migrant MDR clades showed evidence of movement between UN geographic regions (figure 1, table 1A, and online supplementary appendix figure 2) and over 80% (26 of 32) showed evidence of movement between non-adjacent countries, which accounts for migration between non-contiguous countries within the same UN geographic region. While we did not impute the geographic directionality of strain movement, most (73%; 16 of 22) migrant MDR clades having members moving between UN geographic regions indicated migration between countries designated by the WHO as high burden and non-high burden for MDR-TB (table 1A), likely indicating import of MDR-TB from high burden MDR-TB settings into low-incidence countries.
In every case of MDR migration, evidence for migration came from drawing together genomic data from separate studies into one14 (online supplementary appendix figure 3). For example, strains isolated from individuals in Sweden20 belonged to 13 distinct MDR clades that each shared recent MDR ancestry with isolates from patients from 10 other countries, identified across 9 different studies. Thus, by combining data from distinct sequencing efforts in different areas of the globe, we were able to link isolates from patients treated in Sweden to those treated in South Africa, described in Cohen et al;19 in Russia, described in Casali et al;5 in Belarus, described in Wollenberg et al;21 in Moldova, Romania and Iran, described in Manson et al 14 and in other countries, described in Clark et al 22 and Merker et al.11
Supplemental material
Spoligotype analysis revealed that lineage 2 (Beijing) strains, which represented 28% of the strains in our global dataset and were previously documented to have extensive global spread,11 represented more than half (20 of 32) of the observed instances of MDR movement between countries. However, there were also numerous examples involving lineage 4 (Euro-American) strains, which represented 53% of our global dataset—including spoligotypes H, LAM9, T1, T2 and S—and one example involving a lineage 1 (EAI) strain, which represented 9% of our global dataset (see figure 3; table 1A and B). Lineage 4 strains were observed to disseminate among locations in Russia, Eastern Europe, Africa and Sweden, and a lineage 1 clade was observed to have shared descendants in Africa and Sweden.
{kind=link}
{kind=link}
{kind=link}
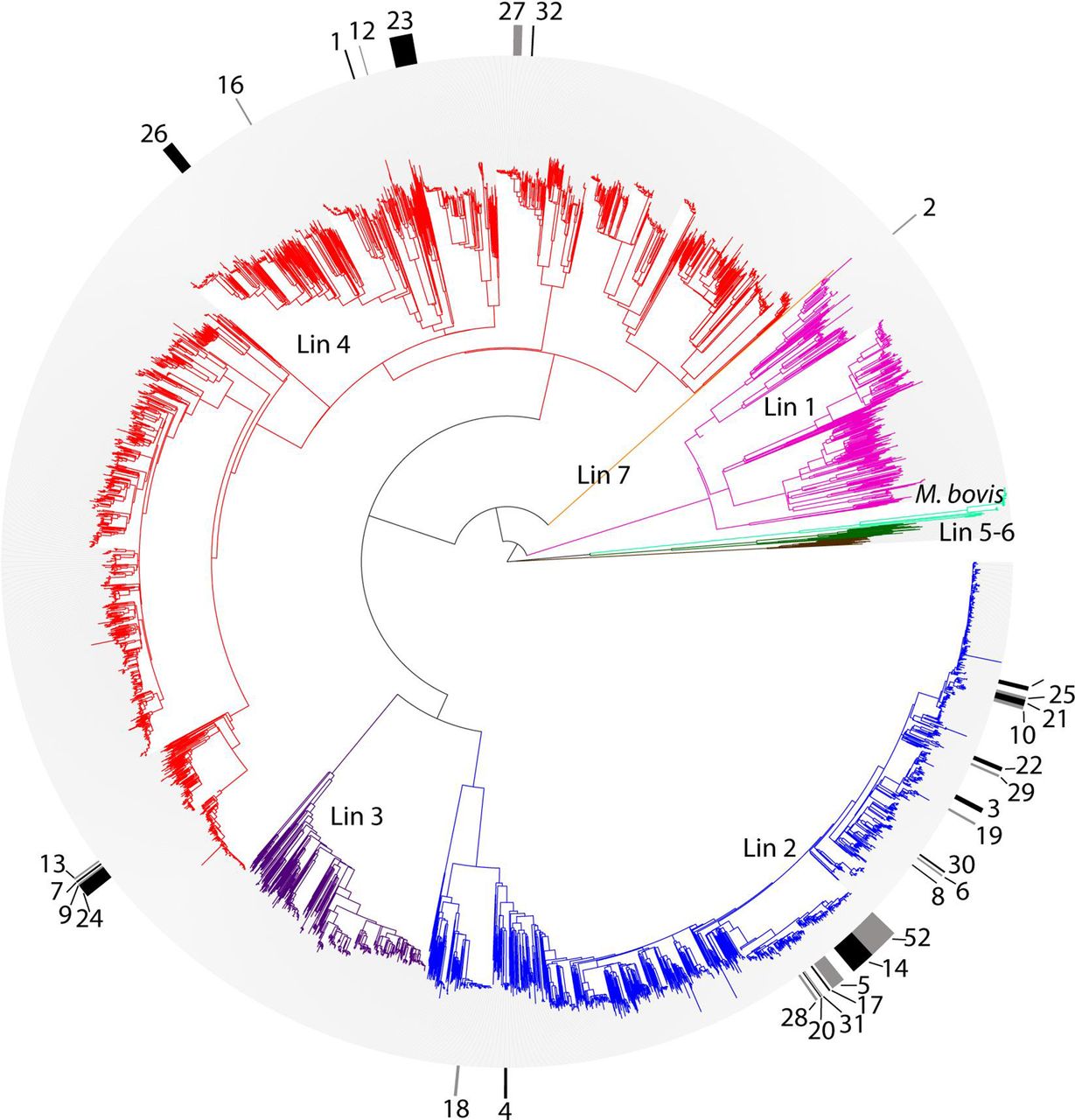
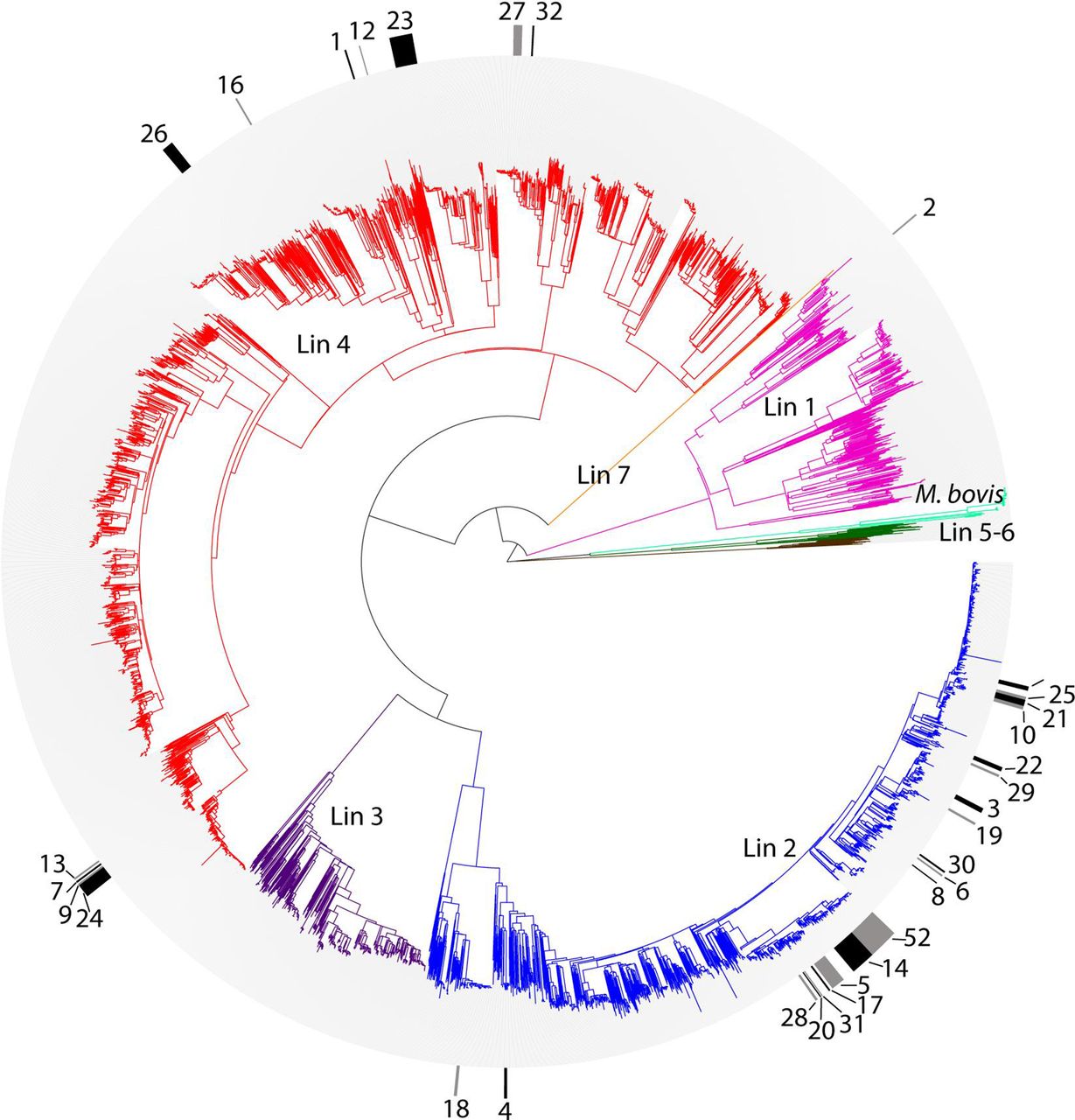
Phylogenetic tree of all 5310 M. tuberculosis isolates. To enhance visualisation of MDR clades on the phylogeny, alternating black and grey markers in the outer circle indicate strains belonging to migratory MDR clades. Clades are numbered per table 1. In the central radial phylogeny, lineages are labelled and colour-coded as follows: pink, lineage 1; blue, lineage 2; purple, lineage 3; red, lineage 4; brown, lineage 5 (M. africanum); dark green, lineage 6 (M. africanum); orange, lineage 7; light green, M. bovis. MDR, multidrug-resistant.
For migrant MDR-TB clades, we calculated the minimum SNP distances between descendant strains isolated in different countries as a proxy for the timing of geographic divergence. For migrant MDR moving between UN geographic regions, the median SNP difference was 17 (range 4–39 SNPs), similar to that calculated for internationally migrant MDR clades (median difference 16 SNPs; range 1–116 SNPs). Assuming a mutation rate of 0.3–0.6 SNPs per genome per year, a difference of four SNPs, the smallest genetic distance between migrant MDR strains we observed, suggests close common ancestry for MDR isolates from patients in Sweden and Uganda, dating back approximately 3–7 years. The median estimate of divergence for migrant MDR-TB clade members was 13–27 years. Thus, whole-genome sequencing can provide both a geospatial and a temporal analysis of the global dissemination of drug-resistant M. tuberculosis strains.
Discussion
In a global dataset of whole-genome sequences from 5310 clinical isolates of M. tuberculosis, we investigated the evolutionary relationships among MDR strains. Combining sequencing data from published studies and disparate global regions allowed us to disentangle de novo evolutions of MDR within a country from person-to-person spread of MDR into a country or region, in a way which could not be done within a single, geographically constrained study. By examining the distinct geographic sites of isolation of all descendants from a single MDR evolution, we saw evidence for 32 internationally migrant MDR clades, of which 22 had migrated between UN geographic regions.
The relationship between the global M. tuberculosis population structure and geographical distribution of strains has been the subject of prior investigation.23–29 Each of the main lineages of M. tuberculosis has been observed to associate with a particular region of the globe.23 Geographic dissemination of M. tuberculosis has been attributed to recent political events, human migration patterns and other ecological drivers of evolution.28–31 Previous sublineage phylogeographic investigations have focused on the widespread dissemination of Beijing (lineage 2) strains,10 11 and the differential geographic restriction of certain lineage 4 sublineages.32 However, to date, few studies have linked phylogeography to global dispersal of MDR-TB or have been unable to find evidence for migration of drug-resistant strains. For example, in a recent global analysis of 1669 lineage 4 M. tuberculosis strains, Brynildsrud et al 26 did not observe drug-resistant strains crossing international borders.
In addition to human migration patterns, global dissemination of M. tuberculosis is affected by the relative fitness of the infecting strains to be transmitted person-to-person and cause active TB disease. Primary transmission of drug-resistant TB, including MDR and XDR has now been well documented in multiple settings.19 33–36 However, it is less well established whether drug-resistant M. tuberculosis strains are more or less transmissible compared with drug susceptible strains. In a transmission network analysis conducted in Malawi, isoniazid resistance was not associated with increased risk of transmission.37 And there is conflicting evidence from household contact studies that have observed both increased and decreased transmissibility of drug-resistant strains, which potentially underscores differences in the relative fitness of different MDR or XDR clades. In a recent 3-year prospective cohort study in Peru, household contacts of MDR-TB patients were approximately half as likely to develop active TB disease when compared with household contacts of drug-susceptible index cases.38 While these data suggest that MDR-TB may be less fit to transmit, smaller studies have shown similar progression to active TB among household contacts of MDR-TB and drug-susceptible TB cases39 and, in a larger retrospective 4-year household contact study of individuals treated for either MDR or XDR in Peru, contacts of XDR patients developed active TB at nearly twice the rate of MDR contacts.40 Though results of studies like these can be conflicting, our results clearly show that MDR-TB is readily transmissible. It would be of interest to determine whether globally transmissible MDR clades are endowed with special features not found in less widely disseminated MDR clades. This kind of analysis would be helped by more comprehensive global databases of M. tuberculosis genomes.
Given that a third of the world’s population is estimated to be infected with M. tuberculosis, undoubtedly our study, focused on only 5310 isolates, highlights only a very small portion of the international movement of MDR-TB and is skewed by larger studies. In fact, the 17 countries from which we observed migratory clade members include some of the most heavily represented countries in our global dataset, including 7 of the 10 most highly sampled countries (Russia, South Africa, UK, Netherlands, Belarus, Sweden and Uganda). Thus, it is likely that with increased sampling, we would see representatives of migratory clades in additional countries, which are currently not as well-sampled. While previous studies have focused on the global dominance of drug-resistant Beijing strains,10 11 we observed 12 examples of non-Beijing MDR clades that disseminated across borders. The large observed fraction of migratory strains from lineage 2 likely reflects the phylogenetic structure of our data set, which contains densely sampled lineage 2 outbreaks, providing more opportunities to observe international spread among Beijing lineages.
We did not see evidence of international movement of XDR-TB within our global dataset. This lack of observed international spread of XDR may reflect the size of our sample collection or that our study methodology was based on genotypic prediction of XDR phenotype, which is imperfect but has been recently improving for second-line drugs.41 However, it is also possible that this may be due to lower fitness or lower transmissibility of these strains. On a more optimistic note, it may be true that, at present, XDR-TB has been relatively geographically contained. To date, we do not have evidence that XDR-TB strains would be unable to disperse globally in a similar way to MDR-TB.
A large number of migrant MDR-TB clades contained strains spanning low-burden and high-burden MDR-TB countries. It is reasonable to assume that MDR most frequently evolves de novo in high-burden countries and is ‘imported’ into low-incidence TB countries. There were several clades in our dataset that fit this paradigm; for example, clade 18 contained a total of five strains, four of which were isolated in South Africa and one in Sweden; and clade 22 contained a total of seven strains, five of which were isolated in Russia and two in Sweden. However, for other clades, disambiguating the most likely directionality of movement was less clear (online supplementary appendix figure 4). For example, clade 4 contained a total of five isolates, two of which were isolated in Russia, one in Moldova and two in Georgia; thus, it is not clear where MDR-level resistance emerged, where it was subsequently transmitted or in which direction-resistant strains migrated along with infected individuals. Thus, due to the complexity of MDR clades spanning multiple countries, as well as long latency periods for TB, we did not report the directionality of MDR transmission in this study. Furthermore, it is worth noting that the transmission events that resulted in the M. tuberculosis strain dispersal patterns depicted in figure 2 may have resulted from multiple intermediary steps—either additional geographic locations or additional human ‘carriers’—that we did not capture in our dataset. Epidemiological data to confirm chains of human transmission or information regarding country of origin and travel history may make these analyses more feasible, but these data were unavailable.
Supplemental material
A known limitation of parsimony-based analyses is that commonly occurring resistance mutations may be interpreted as having evolved once at a more ancestral position on the tree, rather than at multiple recent positions. Thus, this effect of convergent evolution could result in underestimation of the number of independent MDR clades represented in the phylogeny. However, our choice of parameters for the loss versus gain of resistance, involving a higher cost for loss events than gain events14 should minimise this effect, resulting in a conservative prediction of the total amount of MDR migration, predicting a larger number of smaller MDR clades. In agreement with this, most of the SNP distances in our dataset were remarkably small (as few as 1 SNP). One likely exception is a migrant MDR clade from our dataset that included strains from Russia and Belarus separated by 116 SNPs (table 1B). Using SNP difference as a proxy for time, this large SNP difference would place this clade as having emerged more than a hundred years ago, before there were selective antibiotic pressures for the emergence of MDR-TB, which is unlikely. Thus, in this case, it is more probable that MDR evolved convergently in multiple, more recent instances. With a denser sampling of closely related isolates on this part of the phylogeny, one would be able to disambiguate the evolutionary pattern and provide a more accurate portrayal of the geographic divergence of these strains.
Another limitation of our analysis is that, despite being the largest global dataset of M. tuberculosis strains compiled to date, our dataset contained strains from only 48 countries, including 2245 strains (42%) from high-burden MDR-TB countries. Additional strains from more broad geographic regions would allow us to further contextualise MDR-TB evolution and transmission globally. Similarly, only country-level geographic site of isolation was available for the majority of studied strains, which limited our ability to examine geographic transmission within national borders. In addition, if a large dataset of appropriate sample size and geography were collected in a population-based manner, it would become possible to estimate the relative contributions of de novo versus person-to-person transmission of MDR-TB as well as to discern the directionality of transmission.
While multiple online resources such as SITVIT42 and SPOTCLUST43 exist for interrogating geography with DNA fingerprinting data, similar platforms are in their infancy for genomic data, such as ReSeqTB (https://platform.reseqtb.org/).44 However, coordinated efforts like those sponsored by Public Health England (PHE) to scale up routine whole-genome sequencing to include all prospectively identified M. tuberculosis clinical isolates45 should have profound implications for TB control. Beyond drug susceptibility prediction to optimise antibiotic selection, PHE’s undertaking would enable real-time epidemiology since it would include analysis of whole-genome sequencing data, combined with patient metadata including travel history. Comparing PHE data to an international strain database such as ours would enable global contextualisation of the local TB epidemic, including tracking the source of drug-resistant strains. Improved international whole genome sequence databases would inform both the scientific community and public health authorities with more up-to-date information regarding drug-resistant TB outbreaks and international movement of these strains. Inclusion of strains from parts of the globe not included in this study—such as South America, Asia and the Pacific Islands—will also be necessary to provide comprehensive global tracking.
In nearly all cases of MDR clade movement that we detected, strains from different countries were isolated and sequenced as part of separate studies, highlighting the power of combining data across diverse studies. Larger scale whole-genome sequencing studies will be needed to further elucidate global patterns of drug-resistant strain emergence and movement so that international public health efforts to contain drug-resistant TB can be optimally directed.
References
Footnotes
Contributors This study was designed and conducted by KAC, ALM and AE. Analysed the data: KAC, ALM, TA and CAD. Interpreted results: KAC, ALM. Wrote the manuscript: KAC, ALM, AE. Involved in sample acquisition and handling, including oversight of these activities: AME, BWB, SH, SBC. All authors have read the manuscript and confirm that they meet ICMJE criteria for authorship.
Funding National Institute of Allergy and Infectious Diseases Contract No: HHSN272200900018C and Grant Number U19AI110818 to ALM, TA, CAD, BWB, AE; National Heart, Blood, and Lung Institute T32HL007633 and K08HL139994 to KAC and Burroughs Wellcome Fund Career Award for Medical Scientists to KAC. This research used infrastructure resources from the Broad Institute, the Delft University of Technology, Internet2 and SURFnet (the Dutch research and education network), supported under the Enlighten Your Research Global program.
Disclaimer The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.