Article Text

Abstract
Background Uncontrolled lung inflammation is one of the prominent features in the pathogenesis of lung infection- associated acute lung injury (ALI). Microvesicles (MVs) are extracellular nanovesicles that are generated via direct membrane budding.
Methods Bronchoalveolar lavage fluid (BALF) samples were collected from mice with or without intratracheal lipopolysaccharide (LPS) instillation. BALF MVs were characterised and MV-containing microRNA (miRNA) profiles were assessed and confirmed. Secretion and function of MV-containing miR-223/142 (MV-miR-223/142) were analysed in vivo.
Results In BALF, MVs are mainly derived from macrophages in response to LPS. After intratracheal instillation (i.t.) of LPS or Klebsiella pneumoniae, MV-containing miR-223/142 are dramatically induced in both BALF and serum. Mechanistically, miRNA 3′ end uridylation mediates the packing of miR-223/142 into MVs. To investigate the functional role of MV-miR-223/142, we loaded miR-223/142 mimics into unstimulated MVs and delivered them into the murine lungs via i.t. The miR-223/142 mimics-enriched MVs selectively targeted lung macrophages and suppressed the inflammatory lung responses that were triggered by LPS or K. pneumoniae. Mechanistically, miR-223 and miR-142 synergistically suppress Nlrp3 inflammasome activation in macrophages via inhibition of Nlrp3 and Asc, respectively.
Conclusions In the pathogenesis of lung macrophage-mediated inflammatory responses, MV-miR-223/142 secretion is robustly enhanced and detectable in BALF and serum. Furthermore, restoration of intracellular miR-223/142 via vesicle-mediated delivery suppresses macrophage activation and lung inflammation via inhibition of Nlrp3 inflammasome activation.
- microvesicle
- exosome
- microRNA
- bacterial infection
- LPS
- macrophage
- lung injury
Statistics from Altmetric.com
Key messages
What is the key question?
The primary question in this manuscript is to delineate whether bronchoalveolar lavage fluid (BALF) microvesicle (MV) and MV-microRNA (miRNA) cargo are altered after infectious stimuli both in vivo and in vitro. We further investigated whether BALF MV and MV-miRNA cargo can serve as a novel target to develop diagnostic and therapeutic methods.
What is the bottom line?
Intracellular miR-223/142 suppress NLRP3 inflammasome activation in lung macrophages without stimulation. Infectious stimuli lead to 3′ uridylation of miR-223/142 and subsequently a robust release of miR-223/142 via MVs, which occurs along with the NLRP3 inflammasome activation.
Why read on?
Our study suggests circulating MV-containing miR-223/142 potentially serve as a novel diagnostic marker for lipopolysaccharide/Gram-negative bacterial-induced lung inflammation. Moreover, delivery of miR-223/142 via MVs selectively target lung macrophages and provides a novel anti-inflammatory strategy during Gram-negative bacterial infection.
Introduction
Inflammatory responses are a characteristic feature of the host immune response to bacterial infections. As a double-edged sword, inflammatory responses play an essential role in host defense and immune responses against bacterial infections.1 On the other hand, the aims and regulation of inflammatory responses are imprecise and often become a runaway cascade, which can cause collateral damage in tissues.2 Developing a specific biomarker that both reflects the stages of inflammatory responses and can be detected in the circulating blood stream or body fluid is very important. However, currently known biomarkers such as erythrocyte sedimentation rate and C reactive protein are often non-specific.
MicroRNAs (miRNAs) are a class of small non-coding RNAs that are conserved across species.3 miRNAs can enter body fluids through multiple pathways including leakage from dying cells, active secretion from live cells via microvesicles (MVs) and secretion with RNA-binding proteins.4 Since 2007, accumulating evidence has shown that circulating miRNAs serve as biomarkers for cancer development, metastasis and prognosis.5 Emerging data further support that circulating miRNAs may be used as novel diagnostic and therapeutic targets for a variety of non-cancer diseases such as metabolic abnormalities and cardiovascular disorders.6
Extracellular vesicles (EVs) are released by all cells and are ubiquitous in all body fluids, thus carrying a great potential to serve as diagnostic biomarkers and therapeutic targets.7 EVs are now classified into exosomes (Exos), MVs and apoptotic bodies (ABs) based on the size, components and mechanisms of generation.8 In our previous studies, we have demonstrated that EVs transfer miRNAs from the ‘mother’ cells to recipient cells, and that this transfer of their molecular/genetic cargo reprogrammes the function of the recipient cell.8 9 Therefore, EV-containing cargos not only serve as potential diagnostic markers for disease progression but also as novel targets to develop therapeutic agents. Accumulating evidence suggests that miRNAs are selectively incorporated into EVs rather than randomly wrapped in.10 Therefore, the miRNA profiles in EVs potentially reflect the biological status of the parent cells from which the secreted EVs are derived.
Lung infections place a major burden on public health worldwide and are the leading cause of death in the USA.11 Infections caused by Gram-negative bacteria have features that are of particular concern, such as being highly efficient at acquiring antibiotic resistance. The high mortality and morbidity after bacterial infection often result from an imbalance in host defenses between bactericidal and an excessive inflammatory response that leads to tissue damage.12 In this report, we analysed the macrophage-secreted MVs in response to G-negative bacteria or lipopolysaccharide (LPS). In these macrophage-derived MVs, significantly altered miRNA profiles were explored. We attempted to identify the diagnostic and therapeutic targets for profound lung inflammation by analysing the MV-shuttling miRNAs derived from Gram-negative bacteria or LPS-activated macrophages.
Materials and methods
Reagents
LPS from Escherichia coli O111:B4, miR-223-3 p/miR-142-3 p mimics, inhibitors and respective controls were purchased from Sigma-Aldrich. Phosphate-buffered saline (PBS), fetal bovine serum (FBS), RPMI-1640 and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from Gibco. EV-depleted FBS was purchased from System Biosciences. The canonical 3′ end adenylated hsa-miR-223-3 p (5′ UGUCAGUUUGUCAAAUACCCCAAAA 3′) and 3′ end uridylated miR-223 (5′-UGUCAGUUUGUCAAAUACCCCAUUU-3′) were synthesised by Integrated DNA Technologies (IDT). IDT also synthesised all the adapters and primers used in this study, as described below. Lipofectamine 3000 reagent, gentamicin and protease inhibitor cocktail were purchased from Thermo Fisher Scientific. NLRP3 and CD68 antibodies were purchased from Abcam. CD11c, F4/80, Ly-6G/Ly-6C and CD45 antibodies were ordered from BD Biosciences. E-cadherin, ASC, β-actin and CD40L antibodies were ordered from Cell Signaling Technology. K. pneumoniae (strain from American Type Culture Collection (ATCC) No 43186) was provided by Dr Dela Cruz at Yale University School of Medicine.
Bacteria culture
Cultures of K. pneumoniae were grown overnight in Luria-Bertani medium at 37 °C in a rotator at 250 rpm. They were then sub-cultured into fresh medium and grown to mid-log phase. After culturing, bacteria were pelleted and resuspended in PBS. Bacterial concentrations were assessed by serial dilutions. Bacteria count was estimated by OD600 (optical density) and was diluted to final colony-forming unit (CFU) concentrations as needed for each experiment.
Isolation and cell culture of bone marrow-derived macrophages (BMDMs)
BMDMs were isolated as previously described13 and cultured with 30% L929 cells conditioned medium in DMEM complete medium for 7 days before any further experimental procedure. Human THP-1 monocytes were obtained from ATCC and maintained in RPMI-1640 with 10% FBS. THP-1 cells were differentiated into macrophage-like cells for all the experiments by 150 nM phorbol myristate acetate treatment for 24 hours. HEK293T cells were purchased from ATCC and cultured in DMEM with 10% FBS. To reduce the interference of MVs derived from bovine serum, EV-depleted FBS was used instead. All the cells were maintained at 37°C in a humidified atmosphere of 5% CO2–95% air.
In vitro proinflammatory stimulation
For LPS treatment, cultured BMDM were incubated with LPS at a final concentration of 100 ng/mL for the indicated time. For bacterial stimulation, 107 CFUs of K. pneumoniae were added to 105 BMDM for 1 hour. After the incubation, bacteria were removed by washing, and 50 µg/mL of gentamicin was added to kill any remaining extracellular bacteria, as described before.14 Supernatants and cells from treated cultures were harvested following an additional incubation as indicated.
Cloning, transfection and luciferase assay
To identify which target gene might be responsible for miR-223/142-mediated anti-inflammatory effects, we used TargetScan (Release 7.1) to predict both highly and poorly conserved binding targets. The fragments of the Asc and Nlrp3 3′ untranslated regions (UTRs) were amplified from murine complementary DNA (cDNA) by PCR and inserted into the pRL-TK vector containing a cDNA encoding Renilla luciferase and HSV-TK promoter (Promega). QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) was purchased and used for 3′UTRs mutation (mut). The primer sequences used for cloning and mutation are shown in table 1. The plasmid pGL-3 (Promega) coding firefly luciferase was cotransfected and firefly luciferase activity was used as normalisation control. Transfection of miRNA mimics, inhibitors and plasmids was performed using Lipofectamine 3000 according to the manufacturer’s instructions. At 48 hours after transfection, a luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Firefly and Renilla luciferase activities were measured.
Sequences of DNA oligos for cloning, mutation and 3′-Dumbbell-PCR
Animal study
Wild-type (WT) C57BL/6 and Mir223 knockout mice of both genders (8 weeks of age) were obtained from the Jackson Laboratory. To induce lung inflammation, mice were anaesthetised and intubated intratracheally using a blunt-ended feeding needle. 1 µg LPS or 104 CFUs of K. pneumoniae in 50 µL PBS was given through the needle. Mice were euthanised by an aerosolised isoflurane overdose at the indicated time point after instillation. The number of mice used per group was 5 to 8. For hyperoxia-induced acute lung injury (ALI) model, C57BL/6 mice of both genders (8–10 weeks of age) were placed in a Plexiglas chamber maintained at 100% O2 (hyperoxia group) or in a chamber open to room air (room air group) for the indicated time. All the protocols involving animals in this study were approved by the Institutional Animal Care and Use Committee of Boston University.
Preparation and characterisation of EVs
MVs were prepared by using sequential centrifugation protocols described previously.15 Murine bronchoalveolar lavage fluid (BALF) or conditioned media was collected and centrifuged at 300×g for 10 min to remove floating cells. The supernatant was further centrifuged at 2000×g for 20 min to pellet ABs. To isolate MVs, the AB-depleted supernatant was passed through a 0.8-μm-pore filter followed by centrifugation at 16 000× for 40 min. To track the MVs distribution in the lung, PKH26 Fluorescent Cell Linker Kits for General Cell Membrane Labeling (Sigma-Aldrich) was purchased and used according to the manufacturer’s protocol. For transmission electron microscopy (TEM), a commercial kit for TEM imaging was obtained from 101Bio Corporation. The TEM images were taken using a Philips CM120 EM at the Experimental Pathology Laboratory Service Core (Boston University School of Medicine). Nanoparticle tracking analysis (NTA) data were obtained using Nanosight NS500 at the Nanomedicines Characterisation Core Facility (The University of North Carolina at Chapel Hill). To measure the change of MVs secretion in BALF, flow cytometric analysis was performed as described previously.8 Purified MVs were coupled to 10 mL of aldehyde/sulfate latex beads (Thermo Fisher Scientific) for 2 hours. After blocking, the bead-bound MVs were fixed and incubated with antibodies against different biomarkers as indicated.
Preparation and delivery of small RNA-loaded MVs
Electroporation was used to introduce small RNAs, including miR-223/142 mimics and miR-223/142 inhibitors and their respective controls, into MVs as described before.16 Briefly, 100 pmol miRNA mimics or inhibitors or their respective controls were mixed with 100 µg BALF MVs (quantified by protein content). The final volume was adjusted to 100 µL using sterile PBS. The mixture was loaded into the 100 µL Neon Tip and electroporated at 0.5 kV using 10 ms pulse five times using the Neon Transfection System (Thermo Fisher Scientific). To wash the MVs, 900 µL cold PBS was added after electroporation. MVs were pelleted by centrifugation at 16 000×g for 40 min and resuspended in 50 µL PBS. Then, small RNA-loaded MVs were intratracheally instilled into the lung.
Lung inflammation and caspase-1 activity
For cytospin preparations, the cell suspension was cytocentrifuged at 300×g for 5 min using a Shandon Cytospin 4 (Thermo Fisher Scientific). Total inflammatory cell counts in the BALF were determined using a haemocytometer as previously described.17 BALF cells and lung sections were air-dried and stained with PROTOCOL Hema 3 fixative and solutions (Fisher Scientific). The caspase-1 activity in the murine lung tissue was measured using the Caspase-Glo 1 Inflammasome Assay Kit purchased from Promega. The total protein was used as normalisation control.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described.16 Briefly, lung sections were incubated overnight at 4°C with an antibody against mouse CD68 (macrophage marker) or a Ly-6G/Ly-6C antibody (granulocyte marker). After washing, Alexa 488-conjugated secondary antibodies (Thermo Fisher Scientific) were applied. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole). Images of the stained lung sections were visualised and captured using a fluorescence microscope (Axioplan-2, Zeiss), a high-speed 5 megapixel microscope camera (AxioCam, Zeiss) and the software package (AxioVision, Zeiss) with an N-Achroplan 20×/0.45 (Zeiss) objective lens.
RNA preparation, reverse transcription, Real-Time Quantitative Reverse Transcription PCR (qRT-PCR), PCR array and 3′-Dumbbell-PCR miRNeasy Mini Kit (QIAGEN) were used for the purification of total RNA. For miR-223 and miR-142 detection, qRT-PCR was performed using TaqMan PCR kit (Thermo Fisher Scientific) as previously described.18 Hprt1 was used as a normalisation control. To detect the canonical 3′ end adenylated (3′-AAA) or uridylated (3′-UUU) miR-223 and miR-142, 3′-Dumbbell-PCR was used as described before.19 The sequences of adapters and primers used for 3′-Dumbbell-PCR are shown in table 1.
miRNA quantification by the FirePlex assay
Pooled RNA samples from MVs (three groups including PBS, 6 hours and 24 hours after LPS treatment) were provided to Abcam (Cambridge, Massachusetts, USA) and processed to analyse using the FirePlex miRNA Panel—Immunology (Abcam). Results were background subtracted and normalised. A geNorm algorithm has been applied to generate the normalised data. Data are shown as the signals reported in arbitrary units of fluorescence per miRNA target. The signals are proportional to the average amount of fluorescent target bound to the particles. The heat maps were generated using the Firefly Analysis Workbench software.
Western blot analysis and ELISA
Western blot analysis was performed as described previously.20 In brief, cells or MVs were homogenised in Radioimmunoprecipitation assay buffer (RIPA lysis buffer) supplemented with protease inhibitor cocktail. Protein lysates were resolved on SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gels before being transferred to the polyvinylidene fluoride membrane. DuoSet ELISA Kits for mouse interleukin (IL)-1 beta and IL-18 were purchased from R&D Systems to determine the levels of cytokines in BALF according to the manufacturer’s protocol.
Statistical analysis
Data were analysed with SigmaPlot (Systat Software, San Jose, California, USA). Differences between two groups were analysed using the two-tailed unpaired Student’s t-test for variables with a normal distribution or the Mann-Whitney U test for variables without a normal distribution. Differences between three or more groups were analysed using a one-way analysis of variance with a Tukey’s honest significance test for variables with normal distribution or the Kruskal-Wallis one-way analysis followed by pairwise comparisons using Mann-Whitney U tests for those without a normal distribution. Parametric data are presented as mean with SD. Non-parametric data are presented as boxplots showing medians and 25th and 75th percentiles and whisker showing 10th and 90th percentiles. P-value of ≤0.05 was considered to be statistically significant.
Results
BALF MVs are altered in response to LPS
BALF MVs obtained from control or LPS-treated mice were purified and examined using TEM. The captured MVs in both groups have a typical cup shape (figure 1A). The count and size of BALF MVs were determined using NTA (figure 1B,C). A variety of cell markers were analysed on the isolated MVs, including markers of leucocytes (CD45), macrophages (CD68), epithelial cells (E-cadherin) and MVs (CD40L). Notably, the enhanced expressions of CD45 and CD68 were observed in MVs after LPS stimulation (figure 1D). To confirm this observation, we further analysed the BALF MVS using nanoFACS (figure 1E). LPS stimulation increases the percentage of F4/80+/CD11c+ beads from to 32.54% to 61.34% (95% CI 17.17% to 40.43%) (p=0.0004) (figure 1F). We confirmed that LPS induced the MVs that were secreted from lung macrophages.
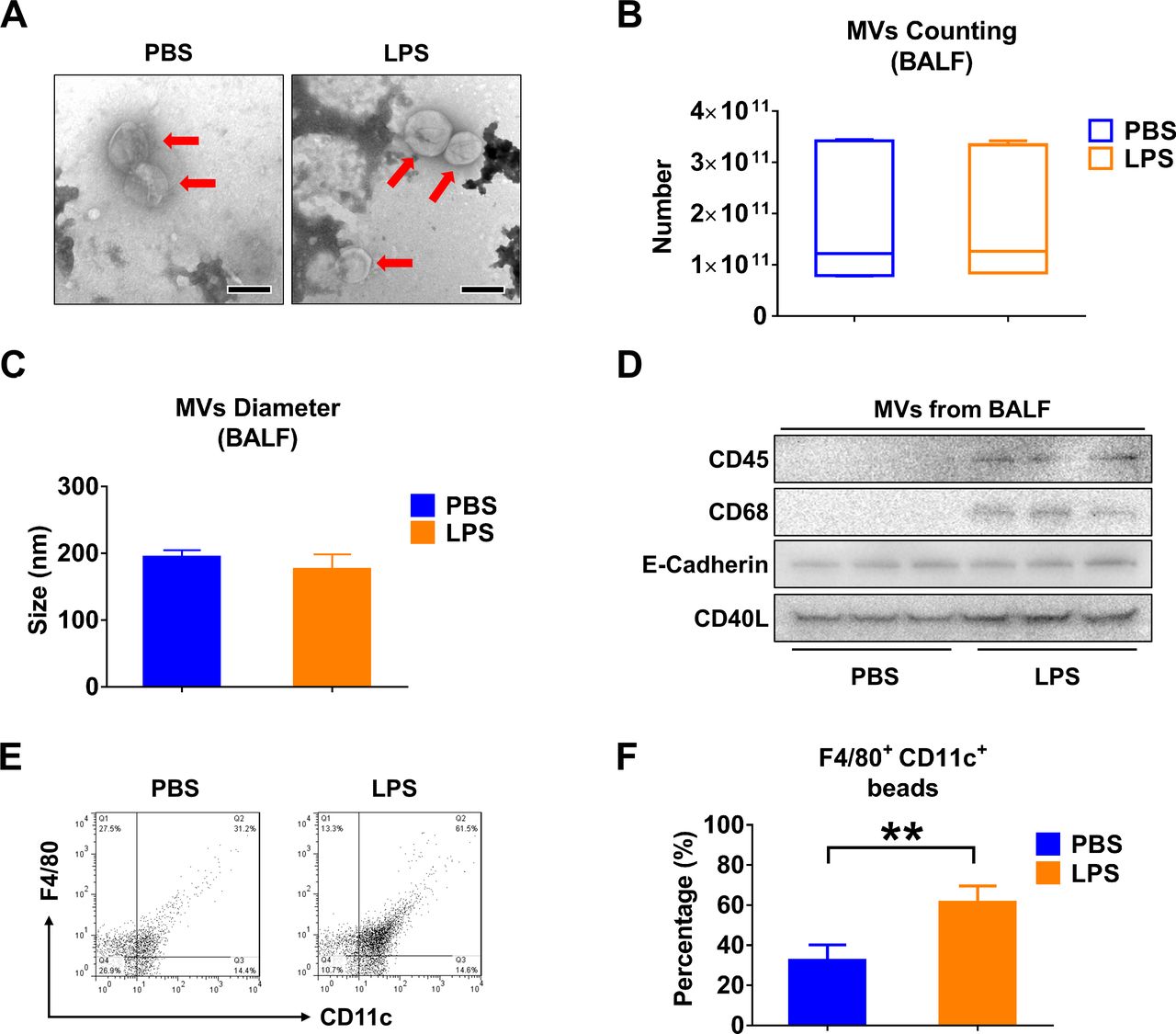
LPS stimulation alters the profile of MVs derived from BALF. Mice received 50 µL PBS or 50 µL PBS containing 1 µg LPS via i.t. After 24 hours, MVs were isolated from BALF. (A) TEM images of BALF-derived MVs were shown, scale bar=200 nm. (B) The number of BALF MVs isolated from mice was counted. The boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles; n=7 mice per group. Data were analysed using a Kruskal-Wallis one-way ANOVA followed by pairwise testing with Mann-Whitney U tests. (C) The diameter of BALF MVs isolated from mice was measured using NTA. Data are mean+SD; n=6 mice per group. Data were analysed using a one-way ANOVA followed by pairwise testing with Student’s t-tests. (D) CD45 (leucocyte marker), CD68 (macrophage marker), E-cadherin (epithelial cell marker) and CD40L (MVs marker) were detected in 100 µg MVs protein using western blot. (E and F) MVs derived from BALF were to flow cytometry analysis (E). The percentage of macrophage-derived MVs (F4/80+CD11c+) in response to LPS stimulation are shown (F). Data are mean+SD; n=5 mice per group. Data were analysed using a one-way ANOVA followed by pairwise testing with Student’s t-tests. **p<0.01 versus the PBS group. ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; LPS, lipopolysaccharide; MVs, microvesicles; NTA, nanoparticle tracking analysis; PBS, phosphate-buffered saline; TEM, transmission electron microscopy.
Infectious stimuli increase miR-223/142 levels in MVs
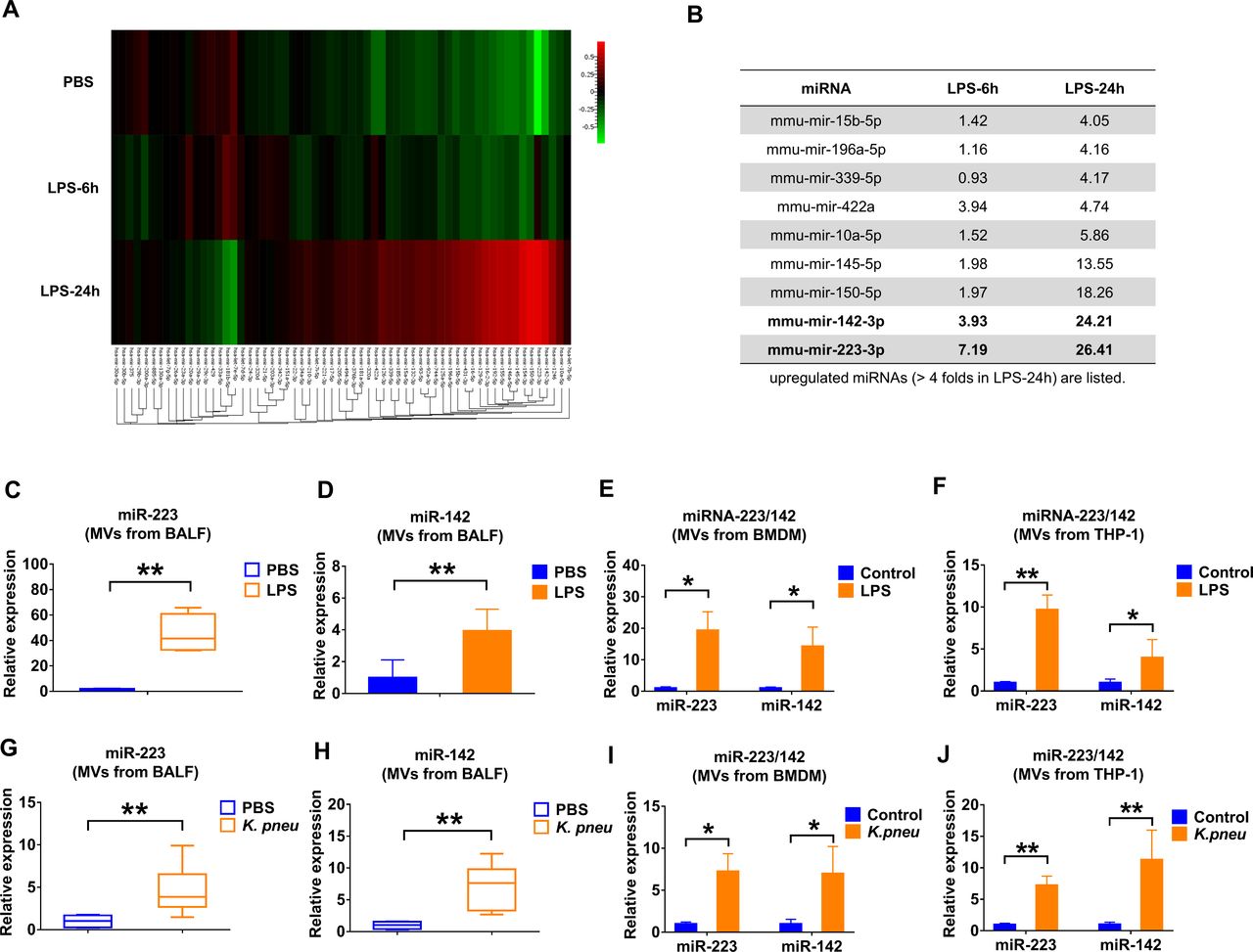
We analysed the miRNA cargo in the isolated BALF MVs using the FirePlex miRNA Immunology Panel (figure 2A). Twenty-four hours after LPS exposure, miR-223 and miR-142 exhibited the greatest induction of expression in MVs, reaching 26.41-fold and 24.21-fold increases compared with controls, respectively (figure 2B). We next validated the enhanced expression of miR-223/142 in MVs using quantitative PCR. In figure 2C, miR-223 in BALF MVs was strikingly upregulated by LPS stimulation with a 47.77-fold increase in expression (p≤0.001). Similarly, miR-142 had a 3.49-fold increase (p=0.002) (figure 2D). Furthermore, the levels of MV-miR-223/142 were detected after LPS treatment in vitro. As shown in figure 2E,F, LPS treatment increases MV-miR-233 derived from both BMDM (19.34-fold, p=0.029) and THP-1 (9.63-fold, p=0.000153), as well as MV-miR-142 secreted from BMDM (14.28-fold, p=0.029) and THP-1 (3.99-fold, p=0.0374). Furthermore, we confirmed the above observations using mice which were exposed to Klebsiella pneumoniae. Consistently, MV-miR-223/142 levels were remarkably elevated both in vivo and in vitro after K. pneumoniae (figure 2G–J).
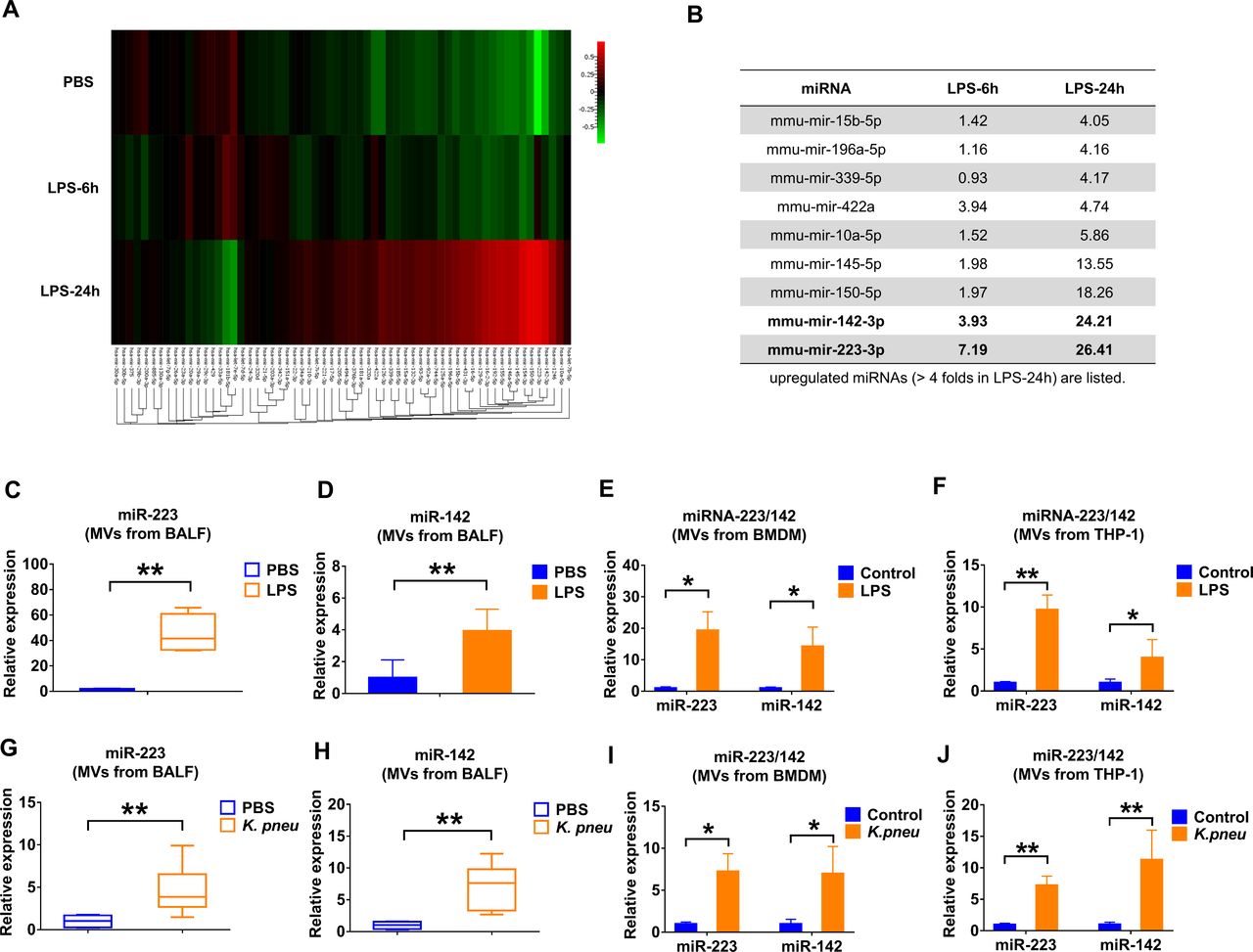
Infectious stimuli elevate miR-223/142 in BALF MVs. (A and B) WT mice (n=7 mice per group) received 50 µL PBS or 50 µL PBS containing 1 µg LPS via i.t. MVs were isolated from BALF at 6 hours or 24 hours after treatment. Pooled RNA from BALF MVs was profiled using the FirePlex miRNA Immunology Panel. The differential expression of miRNAs in MVs from BALF following LPS challenge was shown in the heat map (A). Red and green colours indicate high expression level and low expression level, respectively. Table showing the upregulated miRNAs (B) (fold of increase >4 in MVs derived from BALF, 24 hours after LPS instillation). (C and D) WT mice (n=7 mice per group) received 50 µL PBS or 50 µL PBS containing 1 µg LPS via i.t. After 24 hours, the relative expression of miR-223 (C) and miR-142 (D) in MVs was detected using qRT-PCR. (E and F) MVs were isolated from BMDM (E) and THP-1 (F) culture media with or without LPS treatment (100 ng/mL) for 24 hours. The expressions of miR-223/142 in MVs were detected using qRT-PCR. Results are from three independent experiments. (G and H) WT mice (n=6 mice per group) received 50 µL PBS or Klebsiella pneumoniae (104 CFUs in 50 µL PBS) via i.t. After 24 hours, the relative expressions of miR-223 (G) and miR-142 (H) in MVs were compared using qRT-PCR. (I and J) MVs were isolated from BMDM (I) and THP-1 (J) culture media with or without K. pneumoniae treatment (107 CFU bacteria/105 macrophages, 1 hour incubation). After 24 hours, the expression of miR-223/142 in MVs was detected using qRT-PCR. Results are from three independent experiments. In panels C, G and H, the boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analysed using a Kruskal-Wallis one-way ANOVA followed by pairwise testing with Mann-Whitney U tests. In panels D, E, F, I and J, data are presented as mean+SD and were analysed using a one-way ANOVA followed by pairwise testing with Student’s t-tests. *p<0.05; **p<0.01 versus the PBS or control group, respectively. ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; BMDM, bone marrow-derived macrophage; CFU, colony-forming unit; i.t., intratracheal instillation; LPS, lipopolysaccharide; miRNA, microRNA; MVs, microvesicles; PBS,phosphate-buffered saline; WT, wild type.
3′ End uridylation mediates the package of miR-223/142 into MVs
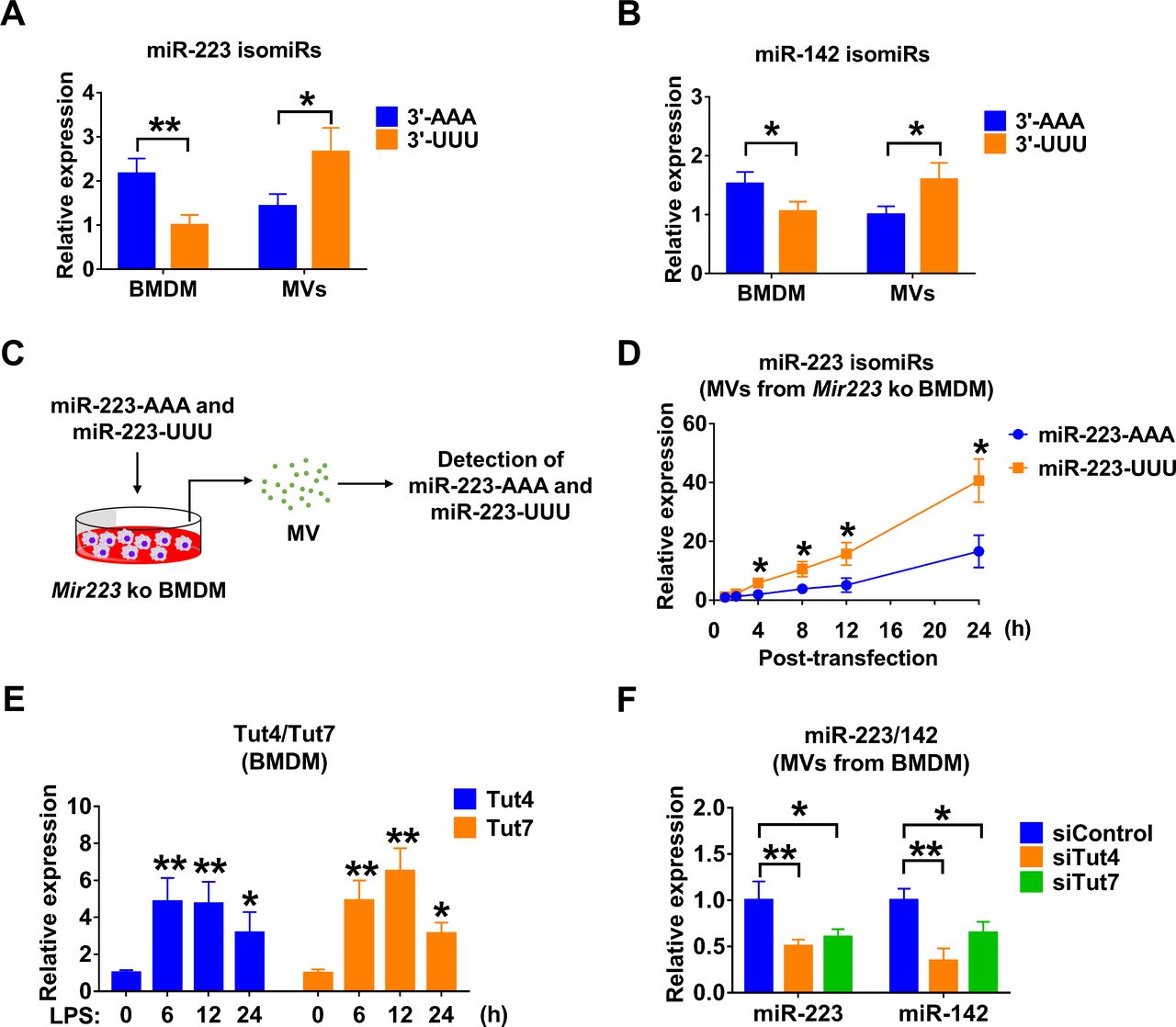
Small RNA compositions in the secreted MVs are different from that in host cells.21 3′ end nucleotide additions have been reported to mediate the selection of miRNAs.21 22 We measured the canonical 3′-AAA or 3′-UUU miR-223/142 in BMDM and BMDM-derived MVs, respectively. We show that 3′-AAA miR-223 was relatively enriched in cells (adenylated : uridylated=2.17:1, p=0.00824), while 3′-UUU miR-223/142 were highly elevated in MVs (adenylated : uridylated=1.43:2.66, p=0.0253) (figure 3A,B). Additionally, we observed that adenylated or uridylated miR-142 has a similar distribution in macrophages and MVs, suggesting that 3′ end nucleotides play a role in the packaging of miR-223/142 into MVs. To further support this hypothesis, we transfected miR-223-depleted BMDMs with synthesised 3′-AAA and 3′-UUU miR-223 (figure 3C). Twenty-four hours after transfection, we found that uridylated miR-223 is significantly more prevalent in secreted MVs than in adenylated miR-223 (adenylated : uridylated=16.60:40.68, p=0.0103) (figure 3D). As previously reported, Tut4 (Zcchc11) and Tut7 (Zcchc6) regulate mature miRNA uridylation.22 Interestingly, we found that LPS dramatically induced Tut4 (4.85-fold, p=0.0010 at 6 hours; 4.74-fold, p=0.002 at 12 hours; 3.16-fold, p=0.039 at 24 hours) and Tut7 (4.91-fold, p<0.001 at 6 hours; 6.49-fold, p<0.001 at 12 hours; 3.12-fold, p=0.024 at 24 hours) in BMDM (figure 3E). In addition, we knocked down Tut4 and Tut7 in BMDM in the presence of LPS using small interfering RNAs (siRNAs). As shown in figure 3F, miR-223 was significantly decreased in MVs released from BMDM transfected with Tut4 (0.50-fold, p=0.009) and Tut7 (0.60-fold, p=0.025) siRNAs. Similarly, MV-miR-142 was decreased after knockdown of Tut4 (0.34-fold, p=0.002) and Tut7 (0.65-fold, p=0.034) (figure 3F).

3' End uridylation mediates the packaging of miR-223/142 into MVs. (A and B) Detection of canonical 3′-AAA or 3′-UUU miR-223 (A) and miR-142 (B) in BMDM and BMDM-derived MVs using 3′-Dumbbell-PCR. (C and D) The experimental design for testing the role of 3′ end adenylation/uridylation-mediated miR-223 packaging into MVs (C). Mir223 depleted BMDMs were transfected with synthesised 3′-AAA or 3′-UUU miR-223. After transfection, MVs were purified from culture media at the indicated time points. The level of adenylated or uridylated miR-223 in MVs was detected using 3′-Dumbbell-PCR (D). (E) BMDMs were stimulated with LPS (100 ng/mL) for the indicated time points. The mRNA expressions of Tut4 and Tut7 were detected using qRT-PCR. (F) BMDMs were transfected with siRNA control, Tut4 siRNA or Tut7 siRNA. After 24 hours, BMDM were treated with LPS (100 ng/mL) for an additional 24 hours. MVs were purified from culture media and subjected to RNA isolation. The expressions of miR-223 and miR-142 in MVs were detected using qRT-PCR. All these results are presented as mean+SD and are from three independent experiments. In panels A and B, data were analysed using a one-way ANOVA followed by pairwise testing with Student’s t-tests. In panels D–F, data were analysed using a one-way ANOVA followed by Tukey’s HSD. *p<0.05; **p<0.01 versus their corresponding control. 3′-AAA, 3′ end adenylated; 3′-UUU, 3′ end uridylated; ANOVA, analysis of variance; BMDM, bone marrow-derived macrophage; HSD, honest significance test; LPS, lipopolysaccharide; MVs, microvesicles; siRNA, small interfering RNA.
Circulating MV-miR-223/142 is a potential marker for lung inflammation
Since BALF MV-miR-223/142 were dramatically elevated in response to infectious stimulation (figure 2), we tested whether circulating MV- miR-223/142 potentially reflect inflammatory lung responses. Interestingly, serum MV-miR-223 were significantly increased in response to LPS (255.34-fold of increase, p=0.007) and K. pneumoniae (320.22-fold of increase, p=0.001) (figure 4A). Meanwhile, serum-derived MV-miR-142 was also upregulated after LPS (68.67-fold of increase, p=0.022) and K. pneumoniae (158.31-fold of increase, p=0.001) treatment. Additionally, the expression of free miR-223/142 was detected in total serum. However, no significant differences were observed between the control group and infectious stimuli-treated groups (LPS and K. pneu moniae) as shown in figure 4C,D. Furthermore, we also performed the FirePlex miRNA Immunology Panel using BALF EVs (figure 4E) and serum EVs (figure 4F) isolated from hyperoxia-induced ALI mice, which is a non-infectious model. As shown in the tables, the miRNA panels are quite different compared with those obtained from infectious models.

Elevated miR-223/142 in serum-derived MVs during lung inflammation. (A and B) Relative levels of miR-223 (A) and miR-142 (B) in serum-derived MVs were detected in control, LPS-treated and Klebsiella pneumoniae-treated mice using qRT-PCR. (C and D) Relative levels of miR-223 (C) and miR-142 (D) in total serum were detected in control, LPS-treated and K. pneumoniae-treated mice using qRT-PCR. The boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles; n=8 mice per group. Data were analysed using a Kruskal-Wallis one-way ANOVA followed by pairwise testing with Mann-Whitney U tests. *p<0.05; **p<0.01 versus the control group. (E and F) C57BL/6J mice (n=6 mice per group) were exposed to room air or hyperoxia (100%). EVs were isolated from BALF or serum after 2 or 3 days as indicated. Pooled RNA from BALF EVs (E) or serum EVs (F) was profiled using the FirePlex miRNA Immunology Panel. The relative fold change of miRNAs was analysed and the upregulated miRNAs in BALF EVs (E) or serum EVs (F) after hyperoxia exposure were shown in tables. ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; EVs, extracellular vesicles; LPS, lipopolysaccharide; miRNA, microRNA; MVs, microvesicles.
miR-223/142 regulate Nlrp3 inflammasome components in macrophages
miR-223 and miR-142 have been identified as anti-inflammatory miRNAs in several studies.23–26 After LPS or K. pneumoniae stimulation, the expression of miR-223/142 in BMDM and THP-1 was decreased while the macrophages were activated (figure 5A–D), indicating that miR-223/142 play an important role in macrophage classical activation in response to infectious stimuli. Furthermore, we identified that NLRP3 and ASC were potential targets of miR-223 and miR-142, respectively (figure 5E). Next, after transfection of miR-223/142 inhibitors or mimics, Nlrp3 (miR-223 mimics: 0.48-fold, p=0.021; miR-223 inhibitors: 1.66-fold, p=0.017) and Asc (miR-142 mimics: 0.44-fold, p=0.047; miR-142 inhibitors: 1.55-fold, p=0.018) protein levels were directly altered, as detected by western blot analysis (figure 5F,G). Dual luciferase assay demonstrated that miR-223/142 mimics were able to repress the 3′ UTR of Nlrp3 (0.29-fold, p<0.001) and Asc (0.43-fold, p<0.001), respectively (figure 5H), and the inhibitory effects were rescued when the predicted miRNA bind site was mutated (figure 5E,H). It is known that NLRP3 and ASC are key components of the NLRP3 inflammasome. Therefore, we then measured the caspase-1 activity in primed BMDMs that were transfected with miR-223/142 mimics. We found a significant inhibition of caspase-1 activity in miR-223/142 overexpressed BMDMs (mimics control: 1 054 931.625±296 326.596 relative luminometer units (RLUs) vs miR-223 mimics: 473 999.250±244 124.986 RLU (p=0.02) vs miR-142 mimics: 512 505.250±253 929.356 RLUs (p=0.029) vs miR-223/142 mimics: 238 909.025±84 816.262 RLUs (p=0.002)) (figure 5I).

miR-223/142 control Nlrp3 inflammasome activity in macrophages. (A and B) Macrophages were cultured with or without LPS (100 ng/mL) for 24 hours. Total RNA was purified from BMDM (A) or THP-1 (B); the expression of miR-223 and miR-142 was detected using qRT-PCR. (C and D) Macrophages were cultured with or without Klebsiella pneumoniae treatment (107 CFU bacteria/105 macrophages, 1 hour incubation). After 24 hours, total RNA was purified from BMDM (C) or THP-1 (D), and the expression of miR-223 and miR-142 was detected using qRT-PCR. (E) Schematic of the miR-223-binding site to the 3′UTR of the NLRP3 and also the miR-142 binding site to the 3′UTR of the ASC. Mutations (underlined) were introduced into NLRP3 and ASC 3’UTR to disrupt base pairing with miR-223 and miR-142, respectively. (F and G) Overexpression and inhibition of miR-223 or miR-142 were performed using transfection. Twenty-four hours later, Nlrp3 (F) and Asc (G) in BMDM were detected using western blot analysis. The relative protein level was quantified and normalised to β-actin. (F) Renilla luciferase reporter containing wild-type or mutant NLRP3 3′UTR was cotransfected into HEK293T cells with miR-223 mimic or mimic control, respectively. Renilla luciferase reporter containing wild-type or mutant ASC 3′UTR was cotransfected into HEK293T cells with miR-142 mimic or mimic control, respectively. Forty-eight hours after transfection, Renilla luciferase activity was measured and normalised to firefly luciferase activity. (G) Mimic control, miR-223 and/or miR-142 mimic was transfected into BMDM. After transfection, BMDM were primed with LPS (1 µg/mL) for 5 min followed by the addition of ATP (5 mM) for 30 min without removal of LPS. Caspase-1 activity in BMDM was measured 24 hours after transfection. All these results presented as mean+SD are from three or four independent experiments. In panels A–D, data were analysed using a one-way ANOVA followed by pairwise testing with Student’s t-tests. In panels F–I, data were analysed using a one-way ANOVA followed by Tukey’s HSD. *p<0.05; **p<0.01 versus their corresponding control. ANOVA, analysis of variance; BMDM, bone marrow-derived macrophage; CFU, colony-forming unit; LPS, lipopolysaccharide; UTR, untranslated region.
miR-223/142 delivery via MVs attenuates the K. pneumoniae-induced lung inflammation
Previously, we developed a protocol using serum-derived Exos as vehicles to deliver designated small RNA molecules into lung macrophages in vivo.16 Here, we slightly modified the method by using BALF MVs. First, we tracked the recipient cells in K. pneumoniae-pretreated mice that took up instilled MVs (figure 6A). Similarly to our previous observation, the majority of PKH26-positive cells were CD68 (green, a marker of monocyte lineage) positive, but Ly-6G/Ly-6C (a marker of polymorphonuclear neutrophils (PMNs)) negative (figure 6B), suggesting that MVs were selectively taken by lung macrophages rather than PMNs.

Delivery of miR-223/142 via MVs attenuates Klebsiella pneumoniae-induced lung inflammation. (A) Schematic illustration of tracking PKH26-labelled MVs after i.t. or delivery of miR-223/142 mimics into K. pneumoniae-pretreated mice via MVs. (B) WT mice received K. pneumoniae (104 CFUs in 50 µL PBS) via i.t. 3 hours before MVs administration. 100 µg PKH26-labelled MVs in 50 µL PBS were given to K. pneumoniae-pretreated mice intratracheally. Mice were then sacrificed 24 hours after MVs administration (n=5 mice per group). Immunofluorescence staining was performed in lung sections using antibodies against CD68 (macrophage marker) or Ly-6G (neutrophil marker) to track MVs uptake. Scale bars=100 µm. (C–H) 100 pmol mimics control or miR-223/142 mimics were electroporated into 100 µg BALF MVs. 50 µL PBS (as a negative control) or small RNA-loaded MVs (in 50 µL PBS) were administrated via i.t. Mice were sacrificed 24 hours after MV administration. The levels of miR-223 and miR-142 were detected in macrophages isolated from BALF (n=5 mice per group) (C). H&E staining was performed using BALF cells and lung sections. Scale bars=100 µm (D) (n=5 mice per group). The number of macrophages or neutrophils collected from BALF was counted (E) (n=6 mice per group). The activity of caspase-1 in murine lung tissue was measured and normalised to total protein (F) (n=6 mice per group). The secretion of IL-1β (G) and IL-18 (H) in BALF was detected using ELISA (n=6 mice per group). In panels C and E, results presented as mean+SD were analysed using a one-way ANOVA followed by Tukey’s HSD. In panels F–H, the boxes in the boxplots show the medians with 25th and 75th percentiles, and the whiskers show the 10th and 90th percentiles. Data were analysed using a Kruskal-Wallis one-way ANOVA followed by pairwise testing with Mann-Whitney U tests. *p<0.05; **p<0.01 versus mimics control group. ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; CFU, colony-forming unit; HSD, honest significance test; i.t., intratracheal instillation; IL, interleukin; miRNA, microRNA; MVs, microvesicles; PBS, phosphate-buffered saline; RLU, relative luminometer unit; WT, wild type.
Next, to evaluate whether miR-223/142-loaded MVs potentially modulate inflammatory lung responses, WT mice were preinfected with K. pneumoniae. After 3 hours, the infected mice received MV-mediated delivery of miRNA mimics control or MV-miR-223/142 mimics intratracheally (figure 6A). The increased levels of miR-223 (3.06-fold of increase, p=0.004) and miR-142 (3.44-fold of increase, p=0.001) were confirmed in macrophages obtained from BALF using qRT-PCR (figure 6C). Significantly less cellular infiltration was observed in lung tissue obtained from the miR-223/142 mimics-treated mice than in the mimics control group (figure 6D). Consistently, total cell counts of macrophages (mimics control: 20.100±3.423×105 mL vs miR-223/142 mimics: 12.150±3.313×105 mL, p=0.014) and neutrophils (mimics control: 95.283±25.674×105 mL vs miR-223/142 mimics: 67.917±13.956×105 mL, p=0.045) were significantly reduced in miR-223/142 mimics-treated mice (figure 6E). Moreover, MV-mediated delivery of miR-223/142 mimics suppressed the inflammasome activity (mimics control: 443.146±153.129 RLU per µg protein vs miR-223/142 mimics: 246.326±81.203 per µg protein, p=0.012) (figure 6F), and secretion of IL-1β (mimics control: 1354.765±479.887 pg/mL vs miR-223/142 mimics: 716.014±337.261 pg/mL, p=0.032) and IL-18 (mimics control: 226.671±41.678 pg/mL vs miR-223/142 mimics: 99.509±16.392, p<0.001) (figure 6G,H) in the lung. Consistently, we observed miR-223/142 inhibitor delivered via MVs promoted LPS induced lung inflammation (data not shown). These data suggested that the delivery of miR-223/142 mimics via MVs promoted anti-inflammatory effect and attenuated the K. pneumoniae-induced lung inflammation.
Discussion
In the past decade, miRNAs have become attractive candidates as novel biomarkers to evaluate human disease development, particularly in the cancer field.27 Compared with protein markers, miRNA levels have unique merits.5 For example, the miRNA level can be measured rapidly and accurately owing to high-throughput sequencing technology.28 Furthermore, the combination of multiple miRNAs which are differentially expressed in different pathways would provide more information than protein markers.29 This is particularly important when developing novel biomarkers using body fluids. Moreover, compared with protein-based markers, detecting miRNAs using PCR and modern sequencing technologies requires much less labour and time than developing a specific antibody against a particular protein. The high stability of miRNAs in body fluids, probably due to their relatively shorter sequences compared with other long non-coding RNAs, represents additional advantages to serve as a biomarker.5 29 Approximately a decade ago, miRNAs were first detectable in circulation within lipid-encapsulated MVs.30 More recently, these lipid-encapsulated MVs have become named EVs. EV-encapsulating miRNAs draw intense interest due to their potential of being developed into non-invasive biomarkers and drug-delivery agents.7 31 The EV cargo is composed of mostly nucleotides and proteins. EVs protect these encapsulated components from degradation.32 Furthermore, EVs carry the same surface protein markers as their originated (parent) cells. This feature can be better used as a biomarker to reflect the pathogenesis of the cell-of-origin.33 Additionally, EVs can serve as a cell-specific carrier to facilitate the delivery of miRNAs as we reported previously.16
It is known that miR-223 and miR-142 belong to haematopoietic tissue-specific miRNAs.34miR-223 and miR-142 were both reported previously to regulate inflammation in tissue and organs other than lungs.23–26 34 The first merit of our studies is that miR-223/142 synergistically suppressed NLRP3 inflammasome activation and bacterial infection-associated lung inflammation in vivo. We chose MVs in our study because miR-223/142 are enriched in MVs when compared with Exos (data not shown). Consistently, previous publications from other groups also show that Exos contain fewer miRNAs.35 More importantly, we found that both miR-223 and miR-142 were selectively secreted from macrophages in an MV-mediated manner. MV is the major type of EVs which are detected after both infectious and sterile stimuli (8,13,15). miRNA 3′ end uridylation was essential in the miR-223/142 encapsulation into MVs. Interestingly, in eukaryotes, uridylation has been reported to alter the half-life of RNA molecules.36 Our data suggest that uridylation of miR-223/142 facilitate the reduction of intracellular levels of miR-223/142, probably via MV-mediated secretion. We added one novel role of uridylation in regulating intracellular levels of miRNAs in response to noxious stimuli.
The significance of the above findings is that MV-miR-223/142 detected in body fluids, including both BALF and serum, are potentially practical biomarkers that reflect the activation of macrophages towards proinflammatory directions. Compared with the detection of non-EV-containing miR-223/142, MV-containing miR-223/142 potentially suggest the specific organs/locations in which macrophages are being activated. MV surface markers are often the same as the surface proteins of their ‘parent’ cells,32 given that MVs are directly generated via surface blebs.37 Furthermore, we do not observe elevated MV-miR-223/142 derived from BALF or serum in hyperoxia-induced ALI mice (figure 4E,F), which is a non-infectious condition. Taken together, our work potentially suggests a novel method by which circulating MV-miR-223/142 can be used to predict lung inflammation and its dynamics postbacterial infections.
Another insight of this current report is that the recipient cells of exogenously delivered MVs are mainly lung macrophages. Based on this feature, we delivered the miR-223/142 mimics in vivo using MVs. Our work confirmed exogenously delivered miR-223/142 prohibited the activation of the NLRP3 inflammasome and provided an anti-inflammation effect in the lung during K. pneumoniae-induced infection. Therefore, MV-mediated miRNA molecules potentially provide a novel therapeutic strategy in a cell-specific manner. Using MVs as a vehicle to deliver exogenous nucleotides has several potential advantages compared with other nanoparticle-mediated delivery methods. First, MVs can be obtained from the blood of the host.38 Therefore, MV-mediated delivery potentially triggers less immune response and may act like the autograft transplantation.39 Next, as stated above, the macrophage-specific MV-mediated delivery illustrated in our report potentially adds more efficacy and less off-target effects when using miRNA molecules as therapeutics.
In our current studies, we compared the levels between MV-miR-223/142 and secreted non-MV-miR-223/142 (figure 4C,D). miRNAs are released in multiple ways, including, but not limited to, a MV-mediated manner. Our data showed that non-MV-shuttling miR-223/142 are not altered in serum. These observations suggested that the detection of the MV-encapsulating miR-223/142 potentially indicates the cellular origin of these released miRNAs. Moreover, we used a systemic inflammation mouse model induced by caecal ligation and puncture (CLP). Total miR-223 level in the serum was detected in both CLP and sham surgery groups. We observed that systemic inflammation induced by CLP increases total miR-233 level in mouse serum, which is different than from lung inflammation (data not shown). Therefore, by detecting the circulating EV-miR-223/142, it might provide a way to distinguish lung inflammation from systemic inflammation.
Currently, we did not address the kinetics of MV-miR-223/142 in our manuscript. MVs are constantly generated and taken up by surrounding cells.40 After noxious stimuli, the secretion of MVs from cells is much more significant compared with their uptake. One of our future directions will focus on addressing the dynamic turnover of MVs and MV-miR-223/142 in our disease models.
As summarised in figure 7, our work demonstrates that miR-223/142 are actively secreted from macrophages via MVs at the time when macrophages are activated toward a proinflammatory direction. Mechanistically, 3′ uridylation plays a role in the miR-223/142 encapsulation into MVs and their secretion into extracellular spaces. MV-miR-223/142 detected in circulation may serve as novel biomarkers to indicate lung macrophage activation and/or lung inflammation. Delivery of MV-containing miR-223/142 mimics in vivo effectively modifies the intracellular level of miR-223/142 and subsequently suppresses macrophage activation via inhibiting NLRP3 inflammasome activation and inflammatory lung responses.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic review of the MVs-mediated secretion and function of miR-223/142 in macrophages during lung inflammation. Infectious stimuli increase in non-templated 3′ uridylation of miR-223/142, which enhances their packaging into MVs. The reduced levels of miR-223/142 via MVs-mediated release in macrophages lead to increased expression of NLRP3 and ASC proteins, two key components of the NLRP3 inflammasome. Accumulated NLRP3 and ASC proteins facilitate the NLRP3 inflammasome activity and promote the maturation and secretion of proinflammatory cytokines IL-1β and IL-18. Besides, miR-223/142-enriched MVs are released into the circulation and might serves as a diagnostic biomarker for lung inflammation. IL, interleukin; MVs, microvesicles.
The table shows the sequences of primers that were used for Asc and Nlrp3 3′UTR cloning/site mutation, as well as 3′-Dumbbell-PCR to detect 3′ adenylation and uridylation of miR-223/142 in the RNA samples as indicated.
References
Footnotes
Contributors DZ, CSDC and YJ designed the research. DZ, HL, LS performed experiments. DZ, HL and XW collected, analysed and interpreted data. DZ, MG and YJ wrote the manuscript.
Funding This work was supported by National Institutes of Health (NIH) grants R21 AI121644, R33 AI121644, R01 GM111313, R01 GM127596, Wing Tat Lee award (all to YJ); by NIH grant K99HL141685 (to DZ).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.