Article Text

Abstract
Rationale Recently a frequent exacerbator phenotype has been described in bronchiectasis, but the underlying biological mechanisms are unknown. Antimicrobial peptides (AMPs) are important in host defence against microbes but can be proinflammatory in chronic lung disease.
Objectives To determine pulmonary and systemic levels of AMP and their relationship with disease severity and future risk of exacerbations in bronchiectasis.
Methods A total of 135 adults with bronchiectasis were prospectively enrolled at three European centres. Levels of cathelicidin LL-37, lactoferrin, lysozyme and secretory leucocyte protease inhibitor (SLPI) in serum and sputum were determined at baseline by ELISA. Patients were followed up for 12 months. We examined the ability of sputum AMP to predict future exacerbation risk.
Measurements and main results AMP levels were higher in sputum than in serum, suggesting local AMP release. Patients with more severe disease at baseline had dysregulation of airway AMP. Higher LL-37 and lower SLPI levels were associated with Bronchiectasis Severity Index, lower FEV1 (forced expiratory volume in 1 s) and Pseudomonas aeruginosa infection. Low SLPI levels were also associated with the exacerbation frequency at baseline. During follow-up, higher LL-37 and lower SLPI levels were associated with a shorter time to the next exacerbation, whereas LL-37 alone predicted exacerbation frequency over the next 12 months.
Conclusions Patients with bronchiectasis showed dysregulated sputum AMP levels, characterised by elevated LL-37 and reduced SLPI levels in the frequent exacerbator phenotype. Elevated LL-37 and reduced SLPI levels are associated with Pseudomonas aeruginosa infection and can predict future risk of exacerbations in bronchiectasis.
- LL-37
- SLPI
- pseudomonas aeruginosa
- elastase activity
Statistics from Altmetric.com
Key messages
What is the key question?
Is there a dysregulation in pulmonary and systemic antimicrobial peptides (AMPs) associated with the frequent exacerbator phenotype in bronchiectasis?
What is the bottom line?
Frequent exacerbators have dysregulation of AMPs with particularly high levels of the cathelicidin LL-37 and low levels of secretory leucocyte protease inhibitor in sputum, which could predict time to next exacerbation and the frequency of exacerbations during follow-up.
Why read on?
This is the first study to describe AMP dysregulation in the frequent exacerbator phenotype in bronchiectasis.
Introduction
Bronchiectasis is a chronic inflammatory lung disease characterised by permanent dilatation of the bronchi. Most patients suffer daily cough and sputum production and some of them experience frequent exacerbations.1 A frequent exacerbator phenotype has recently been described, in which patients consistently have multiple exacerbations over time and this condition independently predicts worse clinical outcomes including increased mortality.2 The underlying biological mechanisms leading to frequent exacerbations in this group have not yet been identified, but frequent exacerbators appear to have greater neutrophilic inflammation and are more susceptible to infection with bacteria, particularly Pseudomonas aeruginosa.3 4
Antimicrobial peptides (AMPs) are important in host defence against pathogenic microbes in the lung.5 Among the most important and abundant AMPs in the airway are lysozyme, lactoferrin and the cathelicidin LL-37 (which are proinflammatory mediators released from activated neutrophils, macrophages and bronchial epithelium) and secretory leucocyte protease inhibitor (SLPI, produced by respiratory epithelial cells and mostly anti-inflammatory).6 7 AMP function through multiple mechanisms including protein degradation (lysozyme), nutrient depletion (lactoferrin), cellular disruption and lysis, and inhibition of virulence factors (LL-37, SLPI).8 9 AMP are designed to protect against bacterial infection but in the context of chronic lung inflammation bacteria such as P. aeruginosa adapt to resist AMP killing, changing methods of iron acquisition and variations in biofilm-associated polysaccharides.10 The result may be an ineffective and excessive AMP response that stimulates inflammation without achieving bacterial clearance.11
A consistent finding in bronchiectasis is the persistence of bacterial infection despite an excessive inflammatory response which instead of achieving bacterial clearance results in host damage.3 12 The contribution of AMP to the vicious cycle of bronchiectasis has not been previously explored. We therefore hypothesised that AMP are altered in sputum from patients with bronchiectasis and may correlate with disease severity and predict future risk of exacerbations.
Methods
Study design and ethics
This was an international, multicentre, prospective, observational study which included consecutive adults with bronchiectasis. The study protocol was approved by local institutional review board and all subjects gave signed informed consent.
Participants
Patients were recruited from three regional specialist bronchiectasis clinics at the Hospital de la Santa Creu i Sant Pau (Barcelona, Spain), Ospedale Maggiore Policlinico (Milano, Italy) and Ninewells Hospital (Dundee, UK).
Bronchiectasis was defined by the presence of bronchial dilatation on high-resolution CT scanning with a compatible clinical history of cough, sputum production and/or recurrent respiratory infections. Patients with cystic fibrosis (CF), primary immunodeficiency, active malignancy, active allergic bronchopulmonary aspergillosis, interstitial lung disease, active mycobacterial disease, HIV infection and long-term oral corticosteroid treatment were excluded.
Clinical assessments
All patients were clinically stable, defined by the absence of an exacerbation that required antibiotic treatment within 30 days. Frequent exacerbators were defined as three or more exacerbations per year at baseline. Demographic data, number of exacerbations in the previous year, relevant comorbid conditions and current treatments were recorded. Exacerbations were recorded as moderate (treated with antibiotics but not requiring hospitalisation) or severe (requiring hospitalisation or intravenous antibiotics). All patients underwent spirometry.13 The underlying aetiology of bronchiectasis was determined after testing recommended by current guidelines.1 14 The Bronchiectasis Severity Index (BSI) and FACED scores were calculated.15 16 Severe disease was considered when BSI score was ≥9 points and FACED score was ≥5 points.
Longitudinal outcomes
Patients were followed up for 1 year from the time of sputum and serum collection. During follow-up, patients were visited every 6 months as part of routine clinical practice at the study centres. Time to first exacerbation was calculated as the time from sampling to the first day of antibiotic administration because of an exacerbation.
Bacteriology
All bacteriology was performed on spontaneous early-morning sputum samples. Sputum was separated from saliva and the sample split for bacteriology and assessment of AMP levels. Samples were processed for bacteriology as previously described.17
AMP and inflammatory mediator measurement
Methods of sputum and blood processing were standardised across the three sites. Sputum samples were processed by ultracentrifugation at 50 000 g for 90 min followed by careful extraction of the supernatant. Samples were processed within 2 hours of expectoration and immediately frozen at −80°C.
Serum and sputum lactoferrin, lysozyme (AssayPro, St. Charles, Missouri, USA), LL-37 (Hycult Biotech, Plymouth, Pennsylvania, USA), SLPI and matrix metallopeptidase 9 (MMP-9) (R&D Systems, Minneapolis, Minnesota, USA) levels were measured by validated commercially available ELISA kits. Sputum samples were diluted 1/25 000 for lactoferrin and lysozyme, 1/20 for LL-37 and 1/2000 for SLPI assays. The limits of detection were 0.625 ng/mL for lactoferrin, 0.0781 ng/mL for lysozyme, 0.14 ng/mL for LL-37 and 0.025 ng/mL for SLPI. Neutrophil elastase (NE) activity in sputum supernatants was measured by activity-based immunoassay (ProAxsis, Belfast, UK) as previously described.18
Western blot
To investigate the degradation of SLPI in bronchiectasis airway sputum, the samples were subjected to denaturing gel electrophoresis on 15% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gels before being transferred to nitrocellulose. After blocking with 5% skimmed milk in phosphate-buffered saline supplemented with Tween 20 (PBST), the membranes were incubated overnight in anti-SLPI antibody at 0.1 µg/mL (catalogue no AF1274; R&D Systems) at 4°C. After incubation with Anti-goat HRP (catalogue no 1721034; BioRad, Watford, UK) at 1/2500 dilution for 1 hour, the blot was washed thoroughly in PBST before SLPI proteins were visualised using Millipore Immobilon Western Chemiluminescent HRP substrate (catalogue no WBKLS0100) used as per manufacturer’s recommendation. A representative autoradiographic film is shown.
Air–liquid interface nasal epithelial cell culture
Nasal basal epithelial cells obtained from healthy subjects and patients with bronchiectasis were grown and seeded into 24-well transwell inserts at p2 or p3. Cells were fully differentiated into ciliated air–liquid interface cultures using Pneumocult ALI+media (Stemcell Technologies, Cambridge, UK). Cells were apically treated with 100 µL/well of phosphate-buffered saline (PBS) (control), NE at 3 µg/mL (Sigma, elastase from human leucocyte, E8140-1UN), with or without elastase inhibitor (AstraZeneca, AZD9668) at 10 nM (a dose sufficient to completely block elastase activity as determined in preliminary experiments) or inhibitor alone. After 30 min of incubation at 37°C, apical supernatants were collected. An additional 100 µL/well PBS was used to wash the apical surface of the cells and added to the corresponding supernatants. Baseline characteristics of healthy controls are described in the Methods section of the online supplementary data.
Supplemental material
Statistical analysis
Statistical analysis was performed using SPSS V.22. Categorical variables are presented as frequencies and percentages and comparisons performed using χ2 test or Fisher’s exact test when required. Continuous variables are presented as mean and SD in parametrical variables and as median and IQR in non-parametrical ones. Differences were analysed using Student’s t-test, analysis of variance test or their corresponding non-parametrical test when required (Kruskal-Wallis and Mann-Whitney U tests). The relationship between linear variables was studied using linear regression and Spearman rank correlation (r and p-value), with the latter included because relationships between inflammatory biomarkers were not assumed to be linear. Data are reported with the r and p-values derived from the Spearman rank correlation. The contribution of different proteases to SLPI levels was studied with multiple linear regression. Exacerbation rate was analysed using a negative binomial model with time in study as an offset. Time to first exacerbation was modelled using Cox’s proportional hazard regression. Exacerbation rate and time to first exacerbation were adjusted for BSI, site and fully adjusted for bronchiectasis severity, gender, aetiology, inhaled corticosteroid use and site. Patients lost to follow-up or dying within the 1-year follow-up were censored in the analysis. Discrimination between groups for the prediction of hospitalisation for severe exacerbations was tested using the area under the receiver operator characteristic curve. A p value <0.05 was considered significant.
Results
Patient characteristics
A total of 135 adults with stable bronchiectasis were included in the study. Table 1 shows the baseline characteristics of the population. Mean age was 69 (±10) years and 56% were females. The majority of patients had idiopathic or postinfective bronchiectasis and the mean BSI score was 8.0 (±4.3) and FACED score was 2.4 (±1.5), indicating moderate to severe bronchiectasis. Most of the patients (65%) were receiving inhaled long-acting bronchodilator treatment, whereas 43% were receiving inhaled corticosteroid treatment and 4% were receiving inhaled antibiotics. Three hundred and twenty-two exacerbations were recorded in the previous year, including 32 (10%) who require hospitalisation. Sixty-eight patients (50.4%) were frequent exacerbators defined as three or more exacerbations per year at baseline. Frequent exacerbators had worse lung function and more severe disease compared with non-frequent exacerbators (see online supplementary table E1).
Patient demographics, clinical characteristics and prior treatments
AMP levels in sputum and blood
All measured AMP in patients with bronchiectasis were higher in sputum than in serum. Lactoferrin was the AMP with highest pulmonary expression, with a median (IQR) of 114.6 (45.7–250.6) µg/mL. In serum, lactoferrin levels were 4.3 (2.3–6.5) µg/mL. Lysozyme was the AMP with highest systemic expression with a median (IQR) of 5.8 (4.4–7.3) µg/mL. In sputum, lysozyme levels were 68.9 (42.1–105.2) µg/mL. LL-37 levels were 1444 (28.1–8054) ng/mL in sputum and 328.3 (204.3–533.1) ng/mL in serum while SLPI levels were 536.4 (161.7–2729) ng/mL in sputum and 120.8 (85.6–152.2) ng/mL in serum.
No correlations between pulmonary and systemic AMP levels were found. Spearman rank correlation between sputum and serum lactoferrin was r=−0.05 (p=0.59), between sputum and serum lysozyme was r=0.06 (p=0.49), between sputum and serum LL-37 was r=0.08 (p=0.36) and between sputum and serum SLPI was r=0.03 (p=0.73).
AMP levels and disease severity
Patients with severe disease had elevated LL-37 levels and lower SLPI levels. Using the BSI score, the highest sputum LL-37 levels were detected in patients with severe BSI score in comparison to patients with moderate and mild disease (3835 (56.9–13 502) vs 108.2 (21.1–6456) vs 148.7 (17.3–2743) ng/mL, p=0.007). Sputum SLPI levels were the lowest in the most severe group in comparison to moderate and mild patients (341.6 (88.8–1635) vs 608.5 (275.9–2678) vs 875.5 (444.1–7415) ng/mL, p=0.004) (figure 1). Using the FACED score, similar relationships were observed to those seen with BSI although overall group differences were not statistically significant (p=0.05 and p=0.1, respectively; see online supplementary figure E1). No correlation among systemic AMP levels and disease severity were found.
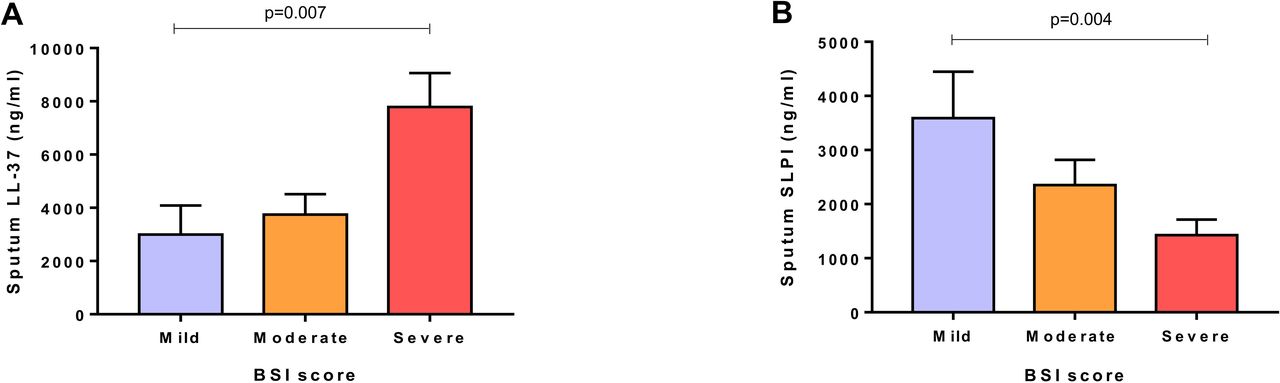
Association of sputum antimicrobial peptide levels and severity of disease. (A) Sputum LL-37 levels and Bronchiectasis Severity Index (BSI) score divided into mild (0–4 points), moderate (5–8 points) and severe (≥9 points). (B) Sputum SLPI and BSI score divided into mild, moderate and severe. P values are obtained by Kruskal-Wallis test. Graphs are represented as mean and SEM.
A weak inverse significant correlation was also observed between sputum LL-37 (r=−0.25, p=0.004), lactoferrin (r=−0.23, p=0.006) and lysozyme (r=−0.19, p=0.02) with FEV1, whereas sputum levels of SLPI had a weak correlation (r=0.17, p=0.05) (see online supplementary figure E2). No correlations between serum determinations and lung function tests were observed.
Frequent exacerbators had significant lower airway SLPI compared with infrequent and non-exacerbators (350.2 (101.7–2583) vs 560.7 (269.2–2424) vs 934.3 (385.1–7206) ng/mL, p=0.01). Sputum lysozyme, lactoferrin and LL-37 were not associated with baseline exacerbation frequency (see online supplementary figure E3). Systemic AMP levels were not associated with exacerbation frequency.
AMP levels and airway infection
Sputum cultures were positive for bacteria in 86 patients (64%) and negative in 49 (36%). P. aeruginosa was the most frequently isolated pathogen in sputum culture (n=39, 29%), followed by Haemophilus influenzae (n=31, 23%), Staphylococcus aureus (n=6, 4%), Escherichia coli (n=3, 2%), Moraxella catarrhalis (n=2, 1%), Stenotrophomonas (n=2, 1%), Streptococcus pneumoniae (n=2, 1%) and Serratia marcescens (n=1, 0.7%). Clinical characteristics of patients grouped according to the presence of P. aeruginosa, other pathogens or negative sputum culture (who were considered non-infected) are showed online (see online supplementary table E2).
When compared with patients infected by other pathogens and non-infected patients, patients with bronchiectasis with P. aeruginosa showed significantly higher sputum LL-37 levels (3835 (72.2–12 435) vs 2425 (36.2–9658) vs 57.8 (17.1–3651) ng/mL, p=0.002), higher sputum lactoferrin levels (190.7 (68.1–364.3) vs 99.2 (40.4–250.6) vs 79.3 (45.7–201.1) ng/mL, p=0.04) and lower sputum SLPI levels (363.3 (103.7–739.7) vs 753.3 (161.7–2624) vs 1066 (309.5–6245) ng/mL, p=0.001) (figure 2). No differences in systemic AMP levels were found between patients infected by P. aeruginosa, patients infected by other pathogens and non-infected patients.

Sputum antimicrobial peptide levels: (A) LL-37, (B) SLPI, (C) lactoferrin and (D) lysozyme, and the presence of airway infection. Patients are divided into non-infected, infected by other pathogens different than Pseudomonas aeruginosa and infected by P. aeruginosa. P values are obtained by Kruskal-Wallis test. Graphs are represented as mean and SEM.
No differences in sputum AMP levels were found when patients with and without chronic macrolide treatment and inhaled corticosteroids were compared. In addition, no differences were found among patients with and without nebulised antibiotics, although the small proportion of treated patients (n=6, 4%) did not exclude a potentially effect (see online supplementary table E3).
AMP and elastase activity
A significant correlation between sputum LL-37 (r=0.36, p=0.002), sputum lactoferrin (r=0.38, p<0.001), sputum lysozyme (r=0.26, p=0.03) and elastase activity was found (see online supplementary figure E4). A previous study identified that in CF SLPI was degraded by NE into lower molecular weight cleavage products,19 while a further study found degradation by MMP-9.20 We hypothesised that proteases activity in the bronchiectasis airway would be responsible for lower levels of SLPI. We found, however, no significant correlation between SLPI and elastase activity (r=−0.09, p=0.46) or MMP-9 (r=−0.13, p=0.2) (figure 3A,B). In multiple linear regression incorporating elastase activity and MMP-9, proteases accounted for a maximum of 23% of the variance in SLPI levels.

(A) Relationship between sputum SLPI and neutrophil elastase activity. (B) Relationship between sputum SLPI and sputum MMP-9 levels. Rho and p-values were obtained from Spearman rank correlation. (C) Western blot demonstrating degradation pattern of SLPI in sputum. Each column represents an individual patient ordered from left to right in terms of increasing sputum neutrophil elastase activity (measured by immunoassay as described in the Methods section). (D) SLPI secretion from nasal epithelial cells of control and patients with BE. (E) SLPI secretion from control (CON1 and CON2) and BE nasal epithelial cells untreated (control buffer) or treated with elastase, inhibitor or a mixture of elastase and inhibitor. BE, bronchiectasis; CP1, cleavage product 1; CP2, cleavage product 2; CP3, cleavage product 3; FL SLPI, full-length secretory leucocyte protease inhibitor; MMP-9, matrix metallopeptidase 9; NE, neutrophil elastase.
By western blot, we identified a heterogeneous pattern of SLPI cleavage with no association between NE activity and SLPI cleavage pattern (figure 3C). Based on previous work that suggest NE can prevent release of SLPI from lung epithelial cells,21 we tested whether exogenous NE inhibited SLPI secretion from primary airway epithelial cells. We observed that nasal epithelial cells from controls and bronchiectasis secreted similar amounts of SLPI (301.6 (167.4–676.4) vs 163.1 (128.8–340.4), p=0.46) (figure 3D). The addition of exogenous elastase decreased the secretion of SLPI to 21.6% (13.8%–37.7%) of the secretion from cells treated with control buffer (p=0.03). The NE inhibitor alone had no significant impact on SLPI release, while the addition of NE and inhibitor still resulted in the inhibition of SLPI release suggesting that this effect was independent of NE protease activity (p=0.028) (figure 3E).
Longitudinal outcomes
One hundred and thirty-two out of 135 patients (98%) completed 12 months of follow-up. During this time, a total of 310 exacerbations have been recorded in 102 patients, including 36 severe exacerbations. The median number of exacerbations per patient was 2.0. In univariate analysis, patients with three or more exacerbations during follow-up had higher levels of LL-37 (2976 (48.8–15 472) vs 416.4 (21.8–7251) vs 61.4 (19.8–2865) ng/mL, p=0.02) and lower levels of SLPI (316.3 (89.9–944.1) vs 753.3 (343.1–3914) vs 1261 (265.2–6521) ng/mL, p<0.001) in comparison to those patients with one or two exacerbations and patients without exacerbations (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Sputum SLPI levels and frequency of exacerbations during 1-year follow-up. (B) Time to next exacerbation. Percentage of patients free of exacerbation using LL-37 1500 ng/mL as a cut-off (HR 1.68 (95% CI 1.13 to 2.50), p=0.01). (C) Sputum SLPI levels and frequency of exacerbations during 1-year follow-up. (D) Time to next exacerbation. Percentage of patients free of exacerbation using SLPI 1000 ng/mL as a cut-off (HR 0.63 (95% CI 0.42 to 0.94), p=0.02). P values in panels A and C are obtained by Kruskal-Wallis test. Graphs are represented as mean and SEM.
In an exploratory analysis, we examined outcomes for patients above and below a cut-off representing the median of the population (to the nearest whole number). Patients with LL-37 ≥1500 ng/mL had a higher frequency of exacerbations (incidence rate ratio (IRR) 1.57, 95% CI 1.04 to 2.37, p=0.02). Patients also had a shorter time to next exacerbation with LL-37 levels above the median for the population (HR 1.68, 95% CI 1.13 to 2.50, p=0.01). These findings persisted after adjusting for BSI and site (see online supplementary table E4). LL-37 showed an area under the curve (AUC) of 0.76 (0.65–0.86), p<0.0001 for predicting hospitalisation (see online supplementary Figure E5).
For SLPI, low levels were not significantly associated with a higher frequency of exacerbations (IRR 0.76, 95% CI 0.50 to 1.51, p=0.1) but did demonstrate a shorter time to next exacerbation when a cut-off of 1000 ng/mL was used (HR 0.63, 95% CI 0.42 to 0.94, p=0.02) (figure 4). After adjusting for BSI and site, a trend to predict the time to first exacerbation was observed (see online supplementary table E4). SLPI showed an AUC of 0.74 (0.62–0.86), p<0.0001 for predicting hospitalisation for severe exacerbation (see online supplementary figure E5).
Discussion
This study demonstrates that airway AMP, especially high LL-37 and low SLPI sputum levels are associated with disease severity, disease activity and airway infection in patients with bronchiectasis. In addition, patients with higher levels of LL-37 and lower levels of SLPI had a shorter time to the next exacerbation and an increased frequency of exacerbations during follow-up. The relationships between LL-37 and SLPI and both prior and future exacerbations suggest that dysregulation of AMP may be one of the mechanisms underlying the frequent exacerbator phenotype in bronchiectasis. Our findings suggested that AMP may be ineffective as an innate host defence mechanism, especially in those bronchiectasis patients with severe disease and with chronic P. aeruginosa infection. This fact may contribute to maintaining a vicious cycle of increased inflammation without eliminating bacterial infection. If further validated in prospective larger studies, LL-37 and SLPI could represent novel biomarkers to predict future risk of exacerbations in patients with bronchiectasis.
AMPs are important in host defence against bacteria, viruses and fungi.5 They are produced by respiratory epithelial cells, neutrophils and macrophages and play an important role in innate lung defence.6 8 Although no previous comprehensive studies of AMP in bronchiectasis have been conducted, our results complement and extend previous observations in other chronic airway diseases such as chronic obstructive pulmonary disease (COPD) and CF. In COPD, increased sputum levels of LL-37 were associated with airflow limitation, health status and exercise tolerance,22 and lower sputum lysozyme and SLPI levels were detected in patients with bacterial colonisation and during acute infectious exacerbations.23 In bronchiectasis, one previous study demonstrated that high levels of sputum LL-37 were detected in patients chronically infected with P. aeruginosa.24 In our study, we demonstrated that elevated sputum LL-37 and reduced sputum SLPI levels were associated with disease severity, worse lung function and the presence of airway infection, especially due to P. aeruginosa. In addition, sputum lactoferrin and lysozyme levels were also associated to worse lung function and sputum SLPI were lower in frequent exacerbators. Local production of AMP in the lung, probably related to chronic airway inflammation, is likely to be the key mechanism in bronchiectasis, since we observed much higher levels in sputum than in serum and no relationship between serum levels and disease severity.
LL-37 is a human cathelicidin produced by neutrophils and epithelial cells in response to pro-inflammatory stimuli including cytokines, pathogen-associated molecular patterns and tissue injury.25 LL-37 displays antimicrobial activity against Gram-positive and Gram-negative bacteria, fungi and viruses, neutralises Lipopolysaccharide (LPS) activity and protects against endotoxic shock.26 In addition, it is involved in the regulation of inflammation, cell proliferation and apoptosis.27 In our study, we demonstrated that high pulmonary levels of LL-37 are associated to disease severity, worse lung function and airway infection. It may seem contradictory that the presence of airway infection and exacerbation risk is associated with higher levels of an AMP that should reduce infection. However, previous studies primarily in CF have demonstrated that the interaction of LL-37 with DNA and with glycosaminoglycan in the inflamed lung impairs the antibacterial function of LL-37 and its ability to neutralise LPS.28 29 LL-37 is, therefore, able to enhance mucus production and promote inflammation while losing its antimicrobial activity.30 These previous findings likely explain the high levels of LL-37 identified in our study and how LL-37 may contribute to poor outcomes.
Bacterial clearance is the primary function of AMP. However, some pathogens, especially P. aeruginosa, have developed different process of adaptation to innate immune clearance mechanisms in the lung, such as creation of biofilms, resistance to inflammasome-mediated clearance and resistance to AMP.11 A recent study evaluating P. aeruginosa populations using whole-genome sequencing in patients with bronchiectasis showed that P. aeruginosa populations adapt by accumulating loss-of-function mutations. This fact leads to changes in phenotypes including different modes of iron acquisition and variations in biofilm-associated polysaccharides.10 These findings may explain why, despite an appropriate host response increasing AMP secretion, P. aeruginosa persists in the lungs and the elevated AMP levels could contribute to perpetuate an excessive inflammatory response, impacting negatively on clinical outcomes.
In contrast to the LL-37 data, in our study sputum levels of SLPI were lower in the most severe patients and in those infected by P. aeruginosa. The main function of SLPI is to protect local tissue from the detrimental consequences of inflammation, as a result of its antiprotease activity and anti-inflammatory properties.7 31 Previous studies have demonstrated decreased levels of SLPI in patients with CF infected with P. aeruginosa due to NE degradation.19 In keeping with the emerging data that CF and bronchiectasis have highly diverging pathophysiology, we found a different relationship in bronchiectasis. SLPI levels were not related to sputum NE activity or MMP-9 levels. SLPI degradation was heterogeneous with multiple cleavage products and was unrelated to elastase activity in sputum. Most patients, even those with high levels of SLPI by ELISA, had evidence of SLPI degradation suggesting that functional SLPI deficiency may be present even in patients with preserved protein levels. Our data suggest that multiple proteases such as matrix metalloproteinases and cathepsins are likely contributing to SLPI deficiency in the airway. However, protease levels only accounted for a maximum of 23% of the variance in sputum SLPI levels, suggesting an important contribution from reduced SLPI production. In addition, our data demonstrate that elastase reduces the release of SLPI from epithelial cells independent of its protease activity, as previous data suggested.21 Other studies have generated a variant of SLPI resistant to degradation by NE.32 However, our data have important therapeutic implications since NE inhibitors that are in development for the treatment of bronchiectasis would not be expected to restore normal SLPI function in view of our findings.
Our study has limitations. First, this study was exploring multiple biomarkers and so we acknowledge the risk of spurious associations as a result of multiple statistical comparisons. We addressed this, however, by looking at baseline data with multiple markers and then focusing our prospective follow-up study on LL-37 and SLPI only, markers which had shown a clear association with disease severity at baseline. The finding of consistent results during follow-up makes spurious associations very unlikely. Our results were consistent, biologically plausible and robust across all studied AMPs. Second, the absence of a control group prevents the comparison of AMP levels of patients with bronchiectasis and healthy subjects. And finally, although our study is the largest of AMP in bronchiectasis, using well-characterised patients in multicentre and international study, it is not possible to clarify if dysregulated AMP levels are markers of disease severity more than a cause of exacerbation, and further studies are needed to better clarify this point. Our study did not explore how sputum biomarkers could be implemented into clinical practice, which may be limited because of the need for sputum processing.
In conclusion, we found that patients with bronchiectasis show dysregulated AMP characterised by elevated proinflammatory LL-37, lactoferrin and lysozyme with reduced anti-inflammatory SLPI. The frequent exacerbator phenotype is associated with increased levels of LL-37 and degradation of SLPI, providing the first biological characterisation of this key phenotype.
References
Footnotes
OS and LP are joint first authors.
Contributors Study design: OS, SA, JDC. Patient recruitment and data collection: OS, LP, AS, GS-C, AR-T, FB, SA, JDC. Performed experiments and sample processing; LP, EC, AS, DC, AS, HRK, MO, SO, SV. Writing the manuscript: OS, LP, JDC. Revising of the manuscript and approval of submission: all authors. Responsible for the overall content as guarantors: OS, JDC.
Funding European Respiratory Society through the EMBARC2 consortium, Instituto de Salud Carlos III (PI18/00311), Fundació Catalana de Pneumologia (FUCAP), Sociedad Española de Neumología y Cirugía Torácica (SEPAR) and a research Grant from Zambon. OS is supported by PERIS. JDC is supported by the GSK/British Lung Foundation Chair of Respiratory Research.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.