Article Text

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal ageing-related disease linked to mitochondrial dysfunction. The present study aimed to determine whether peroxisome proliferator activated receptor gamma co-activator 1-alpha (PPARGC1A, encoding PGC1α), a master regulator of mitochondrial biogenesis, is diminished in IPF and controls pathologic fibroblast activation. Primary human IPF, control lung fibroblasts and fibroblasts sorted from bleomycin-injured mice were used to evaluate the expression and function of PGC1α. In vitro PGC1α manipulation was performed by small interfering RNA knockdown or overexpression. Fibroblast activation was assessed by quantitative PCR, Western blotting, matrix deposition, secreted cytokine array, immunofluorescence and traction force microscopy. Mitochondrial function was assessed by Seahorse analyzer and mitochondria mass and number by flow cytometry, mitochondrial DNA quantification and transmission electron microscopy (TEM). We found that PGC1α levels are stably repressed in IPF fibroblasts. After bleomycin injury in young mice, PGC1α expression drops transiently but then increases prior to fibrosis resolution. In contrast, PGC1α expression fails to recover in aged mice with persistent fibrosis. PGC1α knockdown alone in normal human lung fibroblasts reduces mitochondrial mass and function while enhancing contractile and matrix synthetic fibroblast activation, senescence-related gene expression and soluble profibrotic and prosenescence signalling. Re-expression of PGC1α in IPF fibroblasts ameliorates all of these pathological cellular functions. Pharmacological treatment of IPF fibroblasts with rosiglitazone, but not thyroid hormone, elevated PGC1α expression and attenuated fibroblast activation. The sustained repression of PGC1α and beneficial effects of its rescue in IPF fibroblasts identifies PGC1α as an important regulator of the fibroblast’s pathological state in IPF.
- idiopathic pulmonary fibrosis
- interstitial fibrosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Idiopathic pulmonary fibrosis (IPF) is a fatal disease linked to mitochondrial dysfunction, but specific roles and therapeutic avenues for ameliorating this dysfunction in IPF fibroblasts remain poorly understood.
What is the bottom line?
Our work identifies stable peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) repression as a key driver of fibroblast metabolic dysfunction, profibrotic and prosenescence signalling and fibrogenic activation in IPF.
Why read on?
Therapeutic strategies aimed at reversing PGC1α repression in IPF may be beneficial in the treatment of the disease, and we find that rosiglitazone but not thyroid hormone exerts beneficial effects on PGC1α levels and activation state of IPF fibroblasts.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a devastating age-related disease characterised by abnormal matrix deposition by lung fibroblasts, leading to organ failure.1 Mitochondrial dysfunction is rapidly emerging as a key pathological feature in the development of fibrogenic diseases, including IPF.2–5 In particular, mitochondrial dysfunction in alveolar epithelial cells is now established as a critical alteration that contributes to the initiation and progression of experimental lung fibrosis.6 7 Although mitochondrial alterations have also been reported in fibroblasts isolated from individuals with IPF, including loss of mitochondrial genes and reduced mitochondria size and number,8 9 the molecular mechanisms leading to mitochondrial dysfunction in diseased lung fibroblasts and the functional implications on their fibrogenic activation are not fully understood.
Peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) is a transcriptional co-activator that influences key metabolic pathways, including mitochondrial biogenesis, oxidative phosphorylation and fatty acid metabolism.10 Alterations in PGC1α gene expression have been linked to numerous chronic diseases, including diabetes, hepatitis and kidney fibrosis.11–13 In addition, PGC1α-deficient mice are more susceptible to bleomycin-induced lung fibrosis6 suggesting potential involvement in lung injury and repair. However, the expression of PGC1α in healthy and pathologically activated lung fibroblasts, its roles in controlling fibroblast activation and its responsiveness to recently proposed therapeutic interventions have not yet been investigated.
Here, we aimed to establish PGC1α as a critical modulator of fibroblast activation and connect its sustained gene repression to the mitochondrial dysfunction that has been observed during lung fibrosis progression.
Methods
Detailed methods are provided in the online supplementary file 1.
Supplemental material
Cell culture
IMR-90 embryonic lung fibroblasts (ATCC, Manassas, Virginia, USA), primary human lung fibroblasts (Lonza, Allendale, New Jersey, USA) and primary human lung fibroblasts isolated from patients with IPF or healthy donors were used between passages 3 and 7.
RNA interference
RNA interference was performed using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Waltham, Massachusetts, USA) as described in the online supplementary file 1.
Plasmids and transfection
Transient transfection was performed with pcDNA4 Myc PGC1α by using Lipofectamine p3000 (Thermo Fisher Scientific) as described in the online supplementary file 1.
Immunofluorescence staining
Cells or slides were immunostained by using alpha-smooth muscle actin (αSMA) or PGC1α antibodies as described in the online supplementary file 1.
Traction force microscopy (TFM)
Traction forces were estimated by measuring bead displacement fields on hydrogels and computing corresponding traction fields using TractionsForAll (http://www.mayo.edu/research/labs/tissue-repair-mechanobiology/software), as described in the online supplementary file 1.
Real-time PCR
Total messenger RNA (mRNA) was isolated and the relative gene expression was analysed as described in the online supplementary file 1. Primers used for the quantitative PCR (qPCR) are listed in table 1.
Mouse and human primer sequences for quantitative PCR analysis
Western blotting
Western blotting analysis of protein lysates was performed as described in the online supplementary file 1.
Mouse model of bleomycin-induced lung injury
Two or 18 months old Col1α1-GFP transgenic mouse (FVB strain) were used. Bleomycin (APP Pharmaceutical, LCC Schaumburg, Illinois, USA) or PBS were intratracheally delivered as described in the online supplementary file 1.
Fibrosis evaluation
Hydroxyproline content was measured using a hydroxyproline assay kit (Biovision, Milpitas, California, USA) as described in the online supplementary file 1).
Fluorescence-activated cell sorting (FACS)
Single cells were isolated from mice lungs after bleomycin or vehicle delivery as described in the online supplementary file 1. FACS-sorted fibroblasts were subjected to mRNA isolation, complementary DNA (cDNA) synthesis and qPCR analysis as described in the online supplementary file 1.
Extracellular matrix (ECM) deposition assay
ECM deposition was measured using collagen I and fibronectin antibodies as described in the online supplementary file 1.
Transmission electron microscopy
Number of mitochondria per cell was evaluated by TEM as described in the online supplementary file 1.
Nicotinamide adenine dinucleotide (NAD+) assay
NAD+biosynthesis was analysed using a NAD+assay kit (Cayman, Ann Arbour, Michigan, USA) as described in the online supplementary file 1.
Mitochondrial mass
Mitochondrial mass was determined by FACS-based determination of mitochondrial MitoTracker green probe and by quantification of MT-ATP6 gene as described in the online supplementary file 1.
Mitochondrial respiration
Oxygen consumption rate (OCR) was measured with Seahorse XFe 24 Extracellular Flux Analyxer (Seahorse Bioscience, North Billerica, Massachusetts, USA) as described in the online supplementary file 1.
Isolation of conditioned medium (CM)
Medium was replaced 6 hours after transfections and cells were incubated for 72 hours. CM was collected, centrifuged and immediately used for recipient cells incubation (72 hours) or stored at −20°C for later use.
Cytokine array assays
Cytokine arrays of CM were generated using Proteome Profiler Human XL Cytokine Array Kit (R&D Systems, Minneapolis, Minnesotta, USA) as described in the online supplementary file 1.
Transforming growth factor-β1 (TGF-β1) quantification (ELISA)
TGF-β1 in CM was quantified by using a human Quantikine ELISA kit (R&D Systems) as described in the online supplementary file 1.
Statistical analysis
Individual data points are shown in all plots and represent data from independent mice, cells or biological replicates from cell culture experiments. Depending on the group size, normality distribution was assessed with D’Agostino-Pearson omnibus, Shapiro-Wilk or Kolmogorov-Smirnov normality tests. Variables with normal distribution are summarised as mean and SD, with statistical comparison between two groups performed using Student’s t-test and comparison of more than two groups performed using one-way analysis of variance (followed by Tukey’s post hoc test). Variables with non-normal distribution are summarised as median and IQR, with statistical comparison between two groups performed using non-parametric Mann-Whitney test and comparison of more than two groups performed with non-parametric Kruskal-Wallis test (followed by Dunn’s post-test). All analyses and plots were generated using GraphPad Prism V.6.0 (La Jolla, California, USA) with statistical significance defined as p<0.05.
Results
PGC1α is stably repressed in IPF fibroblasts and transiently repressed in lung fibroblasts during fibrosis and resolution
To directly characterise human disease-relevant changes in PGC1α expression, we compared human IPF derived and healthy control lung fibroblasts. Across donors, we observed a significant decrease in PGC1α transcript (p=0.008) and protein (p=0.002) levels (figure 1A–C). To evaluate changes in PGC1α expression during lung fibrosis initiation, progression and resolution, we employed a Col1α1-GFP transgenic mouse and an experimental model of bleomycin-induced lung injury, in combination with FACS, and measured Ppargc1a transcripts in freshly isolated lung fibroblasts. As shown in figure 1D, the intensity of the green fluorescent protein (GFP)-labelled lung fibroblast population dramatically but transiently increases (days 10–14) following bleomycin exposure and decreases at later time points (30 days), consistent with the self-limiting and resolving nature of this model reported by others.14 FACS analysis of lung fibroblasts (GFP+/CD31−/CD45−/EpCAM-) demonstrated only modest changes (p=0.048 at day 11) in the total population of these cells following bleomycin injury (figure 1F) but confirmed that the proportion of lung fibroblasts with high GFP intensity increased during the early phase after bleomycin-induced lung injury (days 10–21, p=0.0002 and p=0.003, respectively) and gradually diminished over time (p>0.999 at day 30) (figure 1G). Measurement of hydroxyproline content to evaluate collagen deposition in the lungs at 14, 30 and 75 days after injury showed significantly elevated levels at day 14 (p=0.001) and 30 (p=0.003), but with no significant differences between these days. However, hydroxyproline content trended downwards at 75 days compared with day 30 (p=0.11), consistent with the resolving nature of this model (figure 1H). Notably, the changes in relative population of high-GFP fibroblasts correlate with and precede changes in collagen deposition and resolution. In analysing gene expression from the entire population of GFP+ sorted fibroblasts, we observed that Ppargc1a transcript levels appear to be transiently repressed after bleomycin-induced lung injury and spontaneously recovered over time after bleomycin, becoming significantly elevated above control levels by day 30 (p=0.019) (figure 1I).

Peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) is stably repressed in idiopathic pulmonary fibrosis (IPF)-derived fibroblasts and transiently repressed in mouse fibroblasts isolated from the lungs of young, but not old mice, following bleomycin challenge. (A–C) Quantitative PCR (qPCR) and western blotting analysis showing reduced PGC1α expression in IPF-derived fibroblasts (N=5) compared with healthy fibroblasts (N=5). Data passed Kolmogorov-Smirnov normality test and are expressed as mean and SD and analysed using Student’s t-test. (D) Immunostaining of Col1α1-GFP mouse lung following bleomycin exposure showing collagen I staining that overlaps with green fluorescent protein (GFP)-labelled cells. (E) Fluorescence-activated cell sorting (FACS)strategy to isolate fibroblasts from the lungs of young and old mice following bleomycin exposure. (F and G) FACS analysis of whole GFP+/CD31−/CD45−/EpCAM− population in mouse lung. The overall fraction of GFP+ fibroblasts does not dramatically change in young or aged mice following bleomycin (F), but the high GFP+ subset (G), plotted as percentage relative to the total GFP population shown in (F), reveals a transient increase in the lungs of young mice at day 10 and 21 following bleomycin exposure (young sham, N=5; young bleo 11 days, N=6; young bleo 21 days, N=5; young bleo 30 days, N=8; old sham, N=4; old bleo 30 days, N=9). Data did not pass normality test and are expressed as median and IQR and analysed using Kruskal-Wallis test, followed by Dunn’s post-test. (H) Hydroxyproline assay was used to evaluate collagen deposition in the lungs (young sham, N=8; young bleo 14 days, N=7; young bleo 30 days, N=9; young bleo 75 days, N=7). Data passed Shapiro-Wilk normality test and are expressed as mean and DS and analysed using one-way analysis of variance (followed by Tukey’s post hoc test). (I) Gene expression analysis on FACS-sorted total GFP+/CD31−/CD45−/EpCAM− lung fibroblasts shows transient repression of Ppargc1a transcript levels during the early phase after bleomycin-induced lung injury followed by elevation during the resolution phase (day 30). Fibroblasts from the lungs of old mice fail to elevate Ppargc1a transcript levels at day 30 (young sham, N=5; young 11 days, N=5; young 21 days, N=5; young 30 days, N=8; old sham, N=7; old 30 days, N=9). Data passed Kolmogorov-Smirnov normality test and are expressed as mean and SD and analysed using one-way analysis of variance (followed by Tukey’s post hoc test). (H) Relationship between Ppargc1a levels and GFP high population of fibroblasts sorted 30 days post-bleomycin (young 30 days, N=8; old 30 days, N=9).
IPF risk dramatically increases with age,15 and fibrosis resolution in aged mice following lung injury is absent or significantly delayed compared with young animals.16 To evaluate whether fibroblast Ppargc1a gene expression is differently regulated in aged mice compared with young ones, we exposed aged (18 months) Col1α1-GFP mice to bleomycin and evaluated Ppargc1a gene expression in sorted lung fibroblasts at day 30, comparing to our prior analysis of young mice. FACS analysis demonstrated that the total population of GFP+ fibroblasts was similar between young and aged mice (figure 1F) and that the population of high GFP fibroblasts was much more heterogeneous among aged animals, with a substantial number exhibiting a sustained elevated proportion in aged lungs relative to sham (figure 1G), consistent with previous reports of non-resolving fibrosis in aged mice.15 In contrast to the elevated levels of Ppargc1a transcripts observed in young mouse lungs at this time point, Ppargc1a transcript levels in lung fibroblasts from aged mice were more mixed, and did not differ significantly from sham controls (figure 1I). Plotting the relationship between Ppargc1a levels and the Col1-GFP high population of fibroblasts sorted 30 days post-bleomycin revealed a robust non-linear inverse relationship between these outcomes across both young and aged groups (figure 1J). Together these findings establish for the first time that PGC1α is stably repressed in IPF fibroblasts and demonstrate that resolution of lung fibrosis is associated with reversal of PGC1α repression in fibroblasts, suggesting that the loss of PGC1α expression may contribute to persistent fibroblast activation in IPF.
Loss of PGC1α in human lung fibroblasts promotes their fibrogenic activation
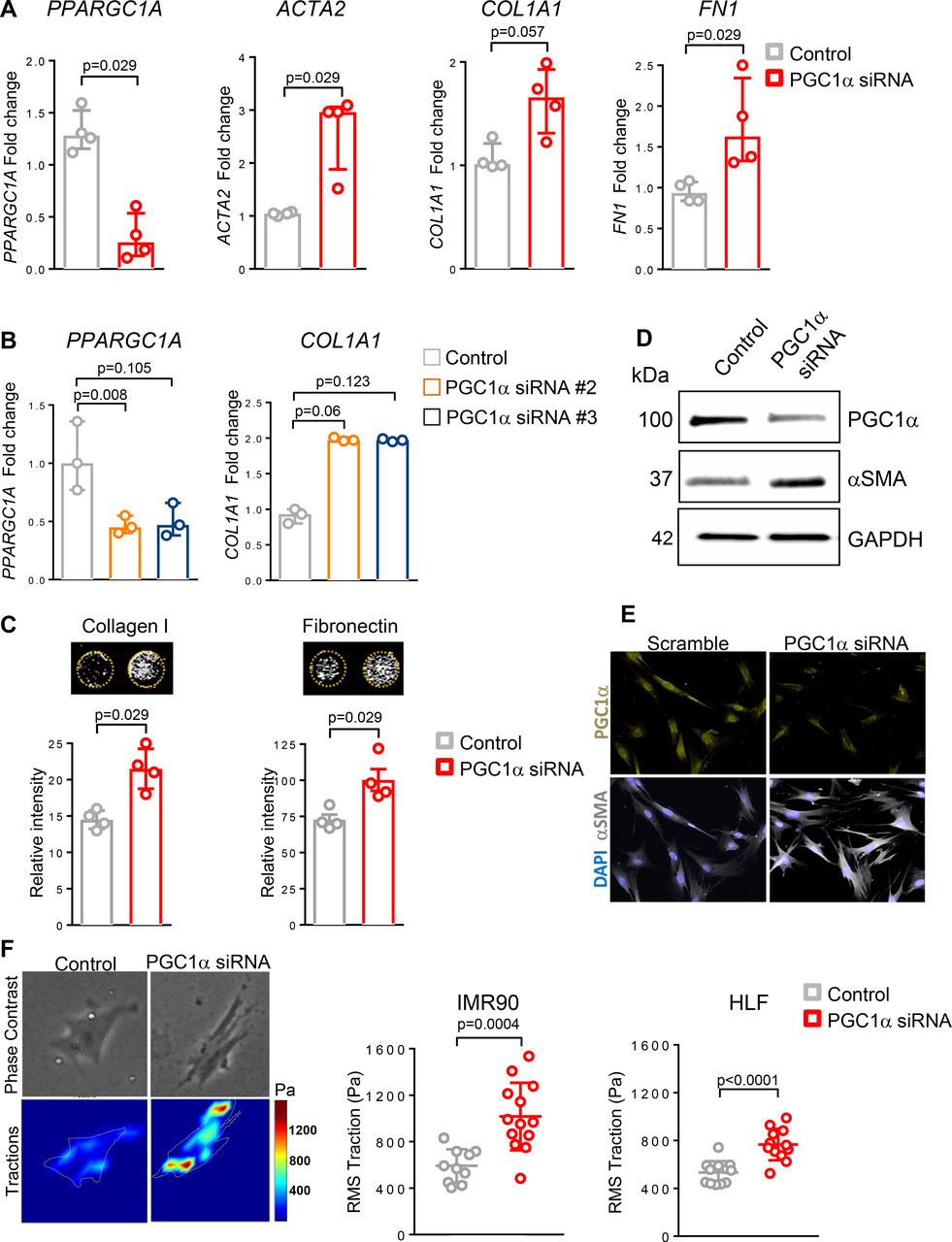
To test whether reduced expression of PGC1α in normal lung fibroblasts directly alters their fibrogenic responses, we used RNAi to knockdown PPARGC1A in primary normal human lung fibroblasts isolated from healthy donor fibroblast (HLF) or fetal human lung fibroblasts (IMR90) for 72 hours. PGC1α silencing in HLF significantly increased profibrotic transcripts, including ACTA2 (p=0.029), COL1A1 (p=0.057) and FN1 (p=0.029) (figure 2A). Similar results were obtained when knockdown was performed using two different small interfering RNA (siRNA) sequences in IMR90 (p=0.06 and p=0.123) (figure 2B) and in HLF (data not shown). To assess de novo matrix synthesis and deposition, we next measured production and deposition of ECM proteins collagen I and fibronectin. As shown in figure 2C, PGC1α knockdown in HLF strongly increased collagen I and fibronectin deposition (p=0.029 for both). To test the effect of PGC1α knockdown on the myofibroblast marker αSMA, we measured its protein expression by western blotting and immunofluorescence imaging. We observed increased αSMA protein using both techniques in PGC1α knockdown cells (figure 2D–E). Using TFM, we confirmed that PGC1α-silenced fibroblasts exert increased forces compared with control transfected cells (figure 2F), directly implicating loss of PGC1α in fibroblast contractile activation (IMR90, p=0.0002; HLF, p<0.0001).

Loss of peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) in normal lung fibroblasts promotes their fibrogenic activation. (A) Gene expression analysis by quantitative PCR (qPCR) demonstrates significant increase in ACTA2, COL1A1 and FN1 transcript levels in PGC1α-silenced normal fibroblasts (N=4 independent experiments). Data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test. (B) qPCR analysis confirms increased COL1A1 transcript levels in PGC1α-silenced fibroblasts using two individual small interfering RNA (siRNA) duplexes (N=3 independent experiments). Data are non-normally distributed and are expressed as median and IQR and analysed using Kruskal-Wallis test, followed by Dunn’s post-test. (C) Extracellular matrix (ECM) deposition assay shows increased collagen I and fibronectin production in PGC1α-silenced lung fibroblasts compared with controls (N=4 independent experiments, each performed in triplicate). Data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test. (D). Western blotting analysis shows alpha-smooth muscle actin (αSMA) elevation in PGC1α-silenced fibroblasts. (E) Representative images of immunofluorescence staining showing increased αSMA expression in PGC1α silenced lung fibroblasts. (F) Traction force analysis demonstrates increased contractility in PGC1α-silenced human lung fibroblasts compared with controls. Representative traction maps are shown (N=2 different cell lines, 10–13 cells analysed for each condition). Data passed D’Agostino-Pearson omnibus normality test and are expressed as mean and SD and analysed using Student’s t-test.
To test whether enhanced expression of PGC1α can reverse these aspects of fibroblast activation, we transfected IPF-derived fibroblasts with a plasmid containing the cDNA encoding for the human PGC1α protein. PGC1α re-expression (72 hours) in IPF-derived fibroblasts significantly reduced ACTA2, COL1A1 and FN gene transcripts (p=0.029 for all) (figure 3A) as well as αSMA protein expression (figure 3B) compared with empty-vector (EV)-transfected cells. Moreover, PGC1α overexpression in these disease-derived cells significantly attenuated both ECM deposition (p=0.029 for both collagen I and fibronectin deposition) (figure 3C) and contractile force generation (p=0.013) (figure 3D).

Peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) re-expression in idiopathic pulmonary fibrosis (IPF)-derived fibroblasts ameliorates their fibrogenic activation. (A) Quantitative PCR (qPCR) analysis of PGC1α-overexpressing IPF fibroblasts (pcDNA4-PGC1α) shows reduced profibrotic transcript levels compared with fibroblasts transfected with an empty vector (EV) plasmid.(B.) Western blotting analysis shows reduced alpha-smooth muscle actin (αSMA) expression in PGC1α transfected IPF fibroblasts compared with EV-transfected control cells. (C.) Extracellular matrix (ECM) deposition assay demonstrates reduced ECM protein deposition in PGC1α overexpressing IPF fibroblasts compared with control fibroblasts (N=4 independent experiments, each performed in triplicate). (D) PGC1α re-expression in IPF lung fibroblasts significantly decreases cell contractility as measured by traction force microscopy. Representative traction maps are shown (N=1 cell line studied, 12 and 7 cells analysed for each condition). (E). qPCR analysis of PGC1α-overexpressing IPF fibroblasts (pcDNA4-PGC1α) shows increased adipogenic genes transcript levels compared with fibroblasts transfected with an EV plasmid (N=3 independent experiments). (F) Nicotinamide adenine dinucleotide (NAD+) determination shows an increased biosynthesis in PGC1α-overexpressing IPF fibroblasts (pcDNA4-PGC1α) compared with fibroblasts transfected with an EV plasmid (N=3 independent experiments, each performed in quintuplicate). Panels A, C, D, E and F: data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test.
PGC1α is linked transcriptionally to PPARy expression,17 which promotes a lipofibroblast fate switch and fibrosis resolution.18 Both PLIN2 (involved in maintenance of adipose tissue19 and lipofibroblasts20) and PPARγ transcript levels were increased in PGC1α-transfected IPF cells (p=0.1 for both) (figure 3E). PGC1α is also linked to NAD+ biosynthesis21 and NAD+ exerts protective roles in both kidney21 and lung fibrosis.22 We observed increased NAD+ biosynthesis in PGC1α-transfected IPF cells (p=0.1) (figure 3F). Together these results demonstrate that loss of PGC1α expression in normal lung fibroblasts promotes fibrogenic and contractile activation, while enhanced expression of PGC1α alone is sufficient to significantly reverse these aspects of IPF fibroblast activation while increasing lipofibroblast and NAD biosynthetic pathways linked to protection from or resolution of fibrosis.
PGC1α expression in human lung fibroblasts controls mitochondrial biogenesis and function
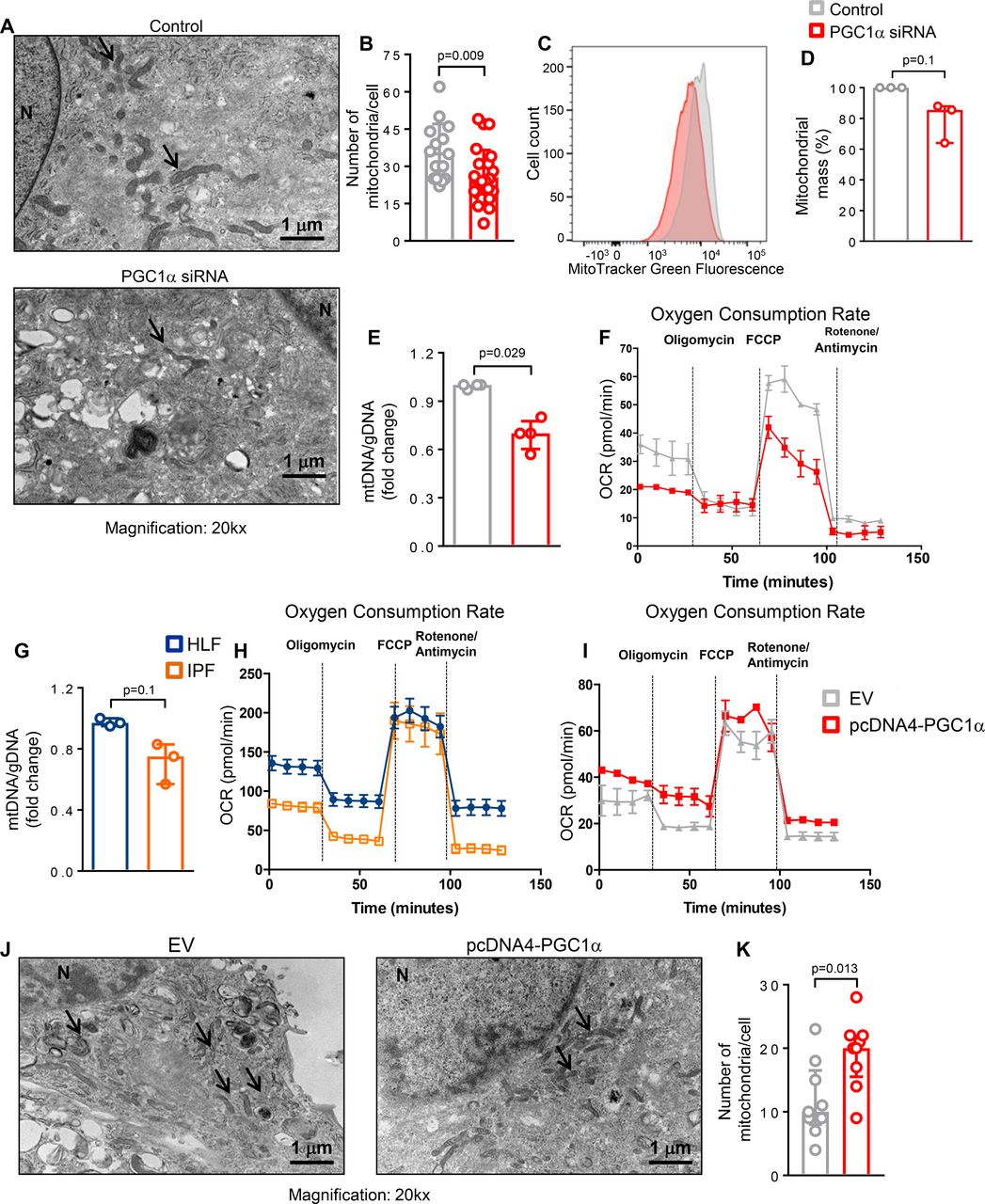
To further assess whether loss of PGC1α in lung fibroblasts affects mitochondrial mass and function, we first evaluated changes in the number of mitochondria using TEM analysis. We found that the number of mitochondria was significantly reduced in PGC1α-silenced human lung fibroblasts 72 hours after transfection compared with control transfected cells (p=0.009) (figure 4A,B). In addition, we compared mitochondrial mass by determining the intensity of MitoTracker Green dye and by quantifying mitochondrial (mtDNA) and genomic DNA. PGC1α-silenced cells exhibited a reduced mitochondrial mass (p=0.1) (figure 4C,D) and ratio of mtDNA to genomic DNA (p=0.029) (figure 4E). To further investigate whether the reduced mitochondrial mass observed in PGC1α-deficient cells leads to altered mitochondrial function, we performed metabolic measurements using a Seahorse analyzer. PGC1α knockdown reduced both basal and maximal mitochondrial respiration (OCR), representing reduced oxidative phosphorylation capacity (figure 4F).

Peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) expression in human lung fibroblasts controls mitochondrial biogenesis and function. (A and B) Representative transmission electron microscopy images and quantification of reduced mitochondria number in PGC1α-silenced human lung fibroblasts (n=24) compared with control cells (n=15). Data passed D’Agostino-Pearson omnibus normality test and are expressed as mean and SD and analysed used Student’s t-test. (C and D) Representative histogram of MitoTracker Green FM fluorescence intensity plotted against number of cells and its quantification showing decreased mitochondrial mass in PGC1α-silenced human lung fibroblasts (N=3 independent experiments). Data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test. (E) Reduced mitochondrial mass in PGC1α-silenced human lung fibroblasts, assessed by mitochondrial DNA (mtDNA)/genomic DNA (gDNA) ratio (N=4 independent experiments). Data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test. (F) Oxygen consumption rate (OCR) bioenergetics profile measured under basal conditions followed by the addition of oligomycin (0.25 µM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCPP) (1 µM) and rotenon/antimycin (1 µM each) shows a reduction of OCR in PGC1α-silenced cells (n=3 independent experiments). (G) Decreased mitochondrial mass (assessed by mtDNA/gDNA ratio) in idiopathic pulmonary fibrosis (IPF)-derived fibroblasts compared with healthy control (N=3 independent experiments). Data are non-normally distributes and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test. (H) OCR bioenergetics profile shows reduced mitochondrial respiration in IPF-derived fibroblasts compared with healthy control (IPF, n=3; HLF, n=2). (I) PGC1α re-expression in IPF cells increases OCR compared with control (n=3 independent experiments). (J and K) PGC1α re-expression in IPF-derived fibroblasts significantly elevates the number of mitochondria compared with empty vector (EV) transfected cells (N=9 for both). Data passed D’Agostino-Pearson omnibus normality test and are expressed as mean and SD and analysed used Student’s t-test.
To explore the relevance of these finding to IPF fibroblasts, we measured the ratio of mitochondrial DNA (mtDNA) to genomic DNA and oxidative phosphorylation in IPF-derived cells (N=3) and control HLF (N=2). Compared with normal lung-derived fibroblasts, IPF fibroblasts exhibited lower mitochondrial mass (p=0.1) and lower basal respiration, although they conserved maximal respiratory capacity (figure 4G,H). Transient overexpression of PGC1α in IPF fibroblasts improved mitochondrial function (figure 4I) and increased mitochondrial number (p=0.029) (figure 4J,K). Collectively, these results demonstrate that reduced PGC1α expression leads to decreased mitochondrial mass and function and that restoring PGC1α in IPF-derived fibroblasts increases mitochondrial number and function.
PGC1α regulates cell autonomous and paracrine fibroblast senescence and fibrogenic signalling
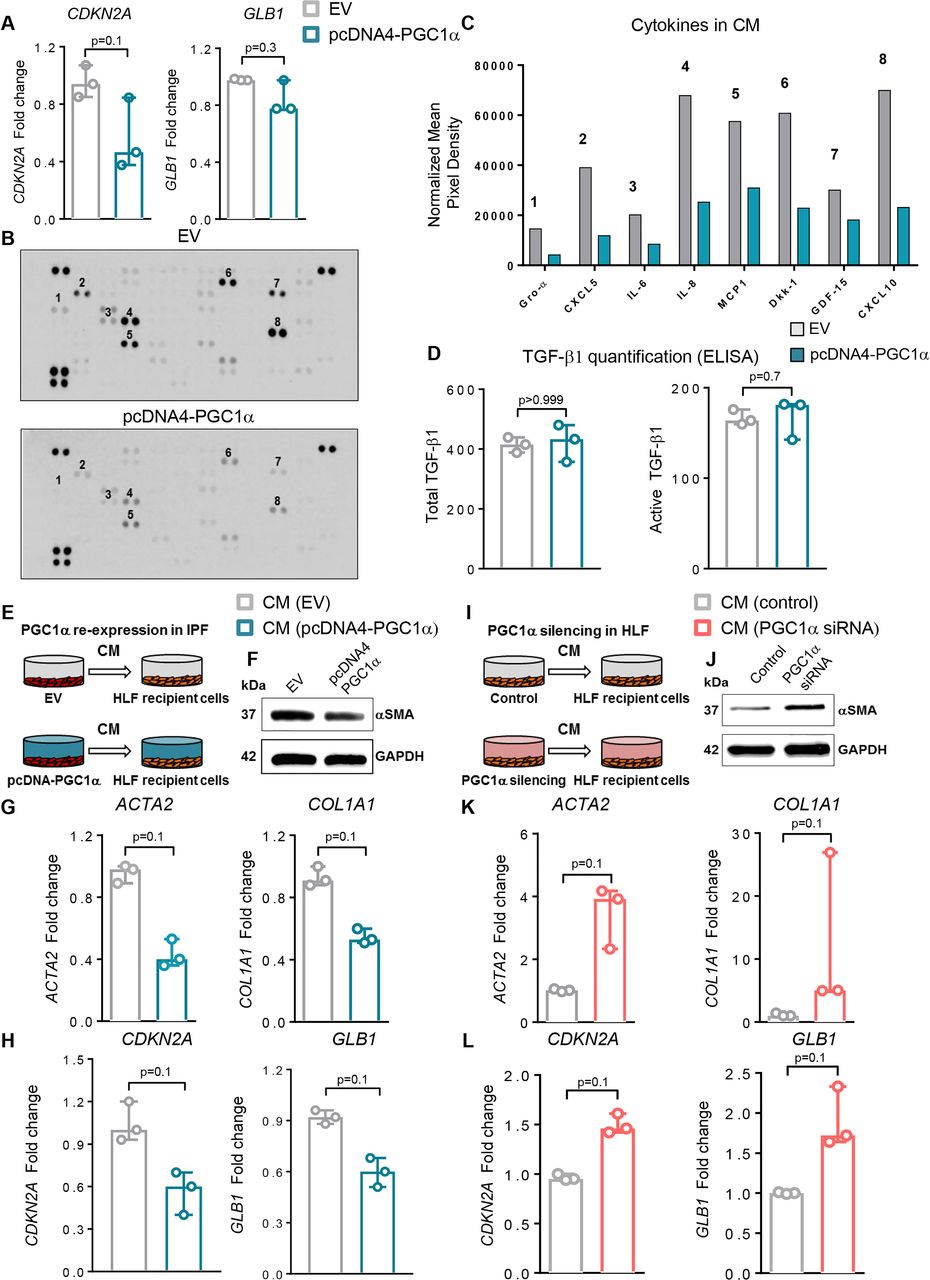
Previous work has linked mitochondrial dysfunction to cellular senescence23 and cellular senescence to IPF.24 25 Senescent cells exhibit a senescence-associated secretory phenotype (SASP) that may contribute to tissue inflammation, wound repair or pathological remodelling26 and may also induce senescence in healthy neighbouring cells in a paracrine fashion.27 More generally, fibroblasts are known to be important sources of soluble signals that influence tissue repair, inflammation and fibrosis.28 To evaluate whether PGC1α regulates expression of senescence markers and fibroblast secretory phenotype, we first measured transcript levels of CDKN2A (p16) and GLB1 (β-Gal) and found that re-expression of PGC1α in IPF-derived cells modestly reduced the expression of these senescence markers (p=0.1 and p=0.3, respectively) (figure 5A). No differences in Caspase 3/7 activity were observed under these conditions, suggesting that the effects of PGC1α expression were independent of apoptosis-related reduction in these senescence markers (data not shown).

Peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) regulates cell autonomous and paracrine fibroblast senescence and fibrogenic signalling. (A) Quantitative PCR (qPCR) analysis of peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α)-overexpressing idiopathic pulmonary fibrosis (IPF) fibroblasts (pcDNA4-PGC1α) shows reduced CDKN2A and GB1 transcript levels compared with fibroblasts transfected with an empty vector (EV) plasmid (N=3 independent experiments). (B) Proteome array analysis of fibrogenic/inflammatory cytokines present in the supernatants of IPF fibroblasts transfected with EV (pcDNA4) or PGC1α plasmids for 72 hours. Conditioned media (CM) from three cultures derived from three independent patients with IPF were pooled and analysed as shown. (C) Relative mean pixel density (normalised to control spots on each membrane) of select proteome array spots in EV transfected and PGC1α re-expressed IPF cells. (D) Quantitative analysis of both total and active transforming growth factor (TGF)-β1 in supernatant of EV or PGC1α-transfected IPF fibroblasts (N=3 independent IPF-derived fibroblasts). (E) Schematic of transfection of IPF fibroblasts prior to CM collection and transfer. CM from EV or PGC1α-transfected cells was collected 72 hours after transfection and transferred to recipient normal healthy donor fibroblast (HLF) for additional 72 hours. (F–G) qPCR (N=3 independent experiments) and Western blotting analysis showing reduced ACTA2 and COL1A1 transcript levels and αSMA reduction in recipient HLF. (H) qPCR analysis shows reduced CDKN2A and GLB1 transcript levels in recipient HLF (N=3 independent experiments). (I) Schematic of transfections of HLF prior to CM collection and transfer. CM from control or PGC1α small interfering RNA (siRNA) transfected cells was collected 72 hours after transfection and transferred to recipient normal HLF for additional 72 hours. (J and K) qPCR (N=3 independent experiments) and western blotting analysis show increased ACTA2 and COL1A1 transcript levels and α-smooth muscle actin (α-SMA) elevation in HLF recipient cells. (L) qPCR analysis shows increased CDKN2A and GLB1 transcript levels in recipient HLF (N=3 independent experiments). Panels A, D, G, H, K and L: data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test.
To test whether PGC1α regulates the release of fibrogenic/inflammatory mediators from these cells, supernatants from EV or PGC1α -transfected IPF fibroblasts (pooled from three independent IPF donor lines) were collected and analysed using a cytokine protein array. The expression of several cytokines was reduced in PGC1α-transfected cells compared with EV control (figure 5B,C), though levels of both total and active TGF-β1 were similar (figure 5D).
The complex nature of the secretory response to PGC1α alterations motivated us to test the integrated effects of these secreted products on naive cells using conditioned media (CM) (figure 5E). PGC1α re-expression in IPF fibroblasts reduced the expression of profibrotic (p=0.1 for both ACTA2 and COL1A1) and senescence markers (p=0.1 for both CDKN2A and GLB1) in naive control lung fibroblasts exposed to conditioned media (figure 5F–H).
To test if the converse is true, we silenced PGC1α in normal fibroblasts and exposed naïve cells to this CM (figure 5I). The CM derived from PGC1α knockdown cells increased expression of profibrotic and senescence markers in naïve control lung fibroblasts (p=0.1 for all) (figure 5J–L). Together these findings demonstrate that PGC1α influences not only the cell-autonomous fibrogenic and senescence signalling state of fibroblasts but also alters their secretory influence on naïve bystander cells. Our findings support the concept that IPF-derived fibroblasts spread their aberrant activation state through soluble signals and that restoration of PGC1α can modulate fibroblast secretory state to attenuate these pathological signals.
Pharmacological elevation of PGC1α by rosiglitazone but not thyroid hormone
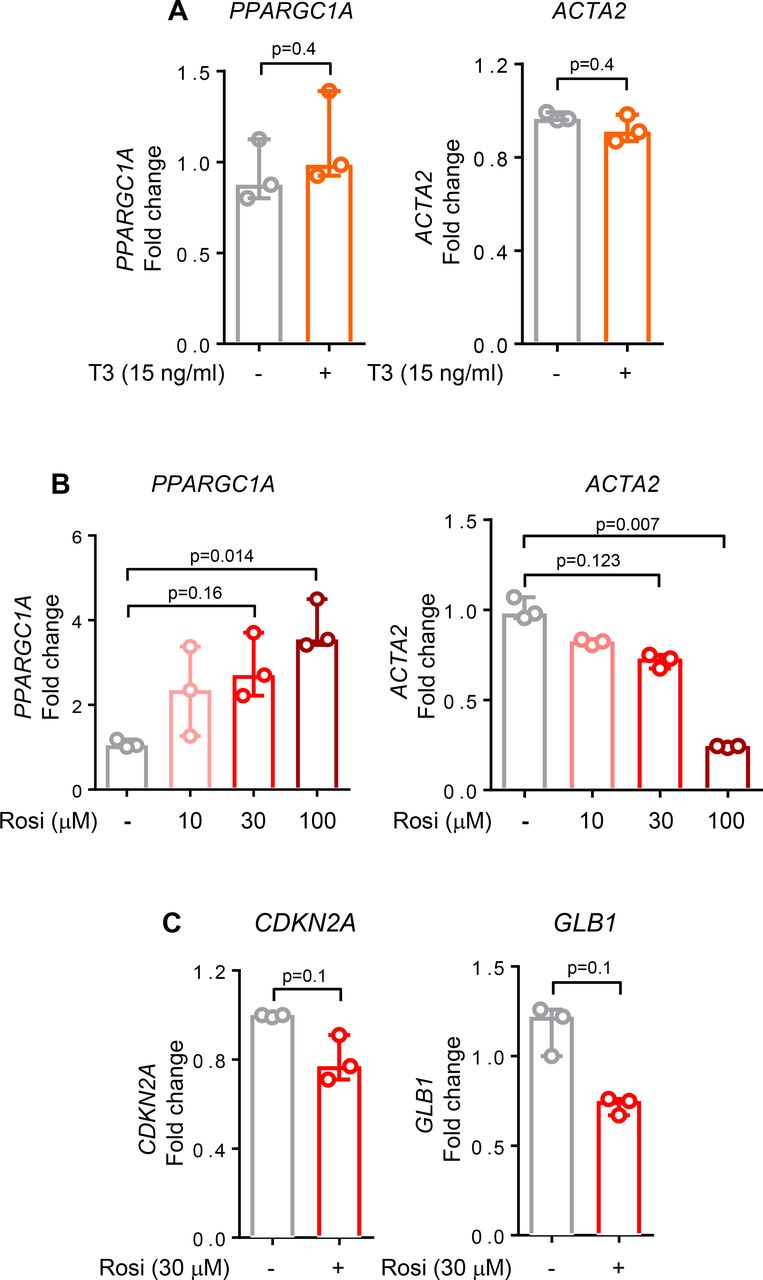
Our findings of PGC1α repression in IPF lung fibroblasts and the beneficial effects of restoring its expression suggest that pharmacological targeting of PGC1α may have therapeutic relevance. Recent work has demonstrated that thyroid hormone (T3) can attenuate experimental lung fibrosis in young mice, specifically by elevating PGC1α expression in alveolar epithelial cells following bleomycin-induced lung injury.6 To test whether similar effects are observed in human IPF-derived lung fibroblasts, we exposed cells to T3 hormone using a dose effective in alveolar epithelial cells.6 Treatment of IPF fibroblasts with T3 hormone failed to elevate PPARGC1A or reduce ACTA2 transcripts in these cells (figure 6A). In healthy lung fibroblasts, T3 hormone treatment alone or after TGFβ stimulation similarly failed to elevate PPARGC1A or reduce ACTA2 transcripts (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pharmacological elevation of peroxisome proliferator activated receptor gamma co-activator 1-alpha (PGC1α) attenuates fibroblast activation. (A) Quantitative PCR (qPCR) analysis of idiopathic pulmonary fibrosis (IPF) cells shows no differences in PPARGC1A and ACTA2 transcript levels after 24 hours treatment with thyroid hormone (T3), 15 ng/mL (N=3 independent experiments). (B) qPCR analysis of IPF cells shows an increase in PPARGC1A and a decrease in ACTA2 transcript levels after 24 hours treatment with rosiglitazone in a dose-dependent manner (N=3 independent experiments). (C) qPCR analysis of IPF cells shows a decrease in both CDKN2A and GLB1 transcript levels after 24 hours treatment with rosiglitazone, 30 µM (N=3 independent experiments). For all panels, data are non-normally distributed and are expressed as median and IQR and analysed using non-parametric Mann-Whitney test.
Rosiglitazone, a PPARγ activator, has antifibrotic effects both in vivo29 and in vitro.30 Rosiglitazone acts through PPARγ dependent and independent mechanisms, including through adenosine monophosphate-activated protein kinase activation,31 a known activator of PPARGC1A gene transcription,32 and exerts beneficial effects in experimental lung fibrosis33 and ageing.34 Rosiglitazone reduced both fibrotic (ACTA2, p=0.007) and senescent features (p=0.1 for both CDKN2A and GLB1) while simultaneously elevating PPARGC1A transcripts in IPF fibroblasts (p=0.014) (figure 6B,C). These findings emphasise that cell-specific targeting may be needed to restore PGC1α function in relevant cell types, including fibroblasts, to achieve therapeutic benefit in IPF.
Discussion
Mitochondrial dysfunction has emerged as an important contributing factor to initiation and maintenance of pathological fibrogenesis in the lungs.5 35 While altered mitochondrial homeostasis leading to dysfunctional responses in epithelial cells is now established as an important player in lung injury and repair, the regulation and contribution of mitochondrial dynamics in fibroblast activation during lung fibrosis remains less explored. Here we demonstrate that the mitochondrial biogenesis regulator PGC1α is transiently repressed in fibroblasts during reversible fibrosis in mice and stably repressed in human IPF fibroblasts. Our in vitro experiments demonstrate that PGC1α plays a critical role in regulating lung fibroblast behaviour and that perturbations in its expression have a profound impact on fibroblast metabolism, matrix deposition, contraction and secretory state.
In young mice, the fibrotic response to injury is typically self-limiting and reversible.14 In aged mice, resolution of fibrosis is dramatically impaired.15 By combining FACS sorting and gene expression analysis in mouse lung fibroblasts, we demonstrated that Ppargc1a expression is dynamically regulated following bleomycin-induced lung injury, with gene repression during injury and fibrogenic phases followed by a significant elevation during the resolution phase. Interestingly, in vitro studies revealed that loss of PGC1α in normal human lung fibroblasts is sufficient to recapitulate multiple features observed in fibroblasts following lung injury including increased contractility, enhanced ECM protein deposition and altered mitochondrial metabolism. These results suggest that transient repression of PGC1α in lung fibroblasts following lung injury may promote fibroblast activation in support of wound healing and that spontaneous recovery of PGC1α expression may be needed to cease the fibrotic response to allow resolution of lung tissue repair. Interestingly, while Ppargc1a expression in fibroblasts spontaneously increases during the resolution phase in young mice, it fails to do so in fibroblasts isolated from the lungs of aged animals exposed to bleomycin. Previous reports have shown that while the magnitude of the early injury responses is comparable between young and old mice treated with bleomycin, old mice experience persistent fibrosis and impaired resolution.16 Thus, our results suggest that the impaired transcriptional reactivation of Ppargc1a in fibroblasts from old mice may contribute to sustained and irreversible lung fibrogenesis. These findings are highly relevant to human disease, as fibroblasts isolated from patients with IPF exhibited stable repression of PGC1α levels. Moreover, metabolic measurements demonstrate that oxidative phosphorylation is altered in IPF fibroblasts, and PGC1α re-expression in these cells restores this energetic imbalance. Notably, previous reports showed that telomere dysfunction is also linked to repression of PGC1α and reduction of mitochondrial biogenesis and function in diverse organs and that restoring PGC1α improve mitochondrial function in the setting of telomere.36 More broadly, PGC1α re-expression in IPF fibroblasts reduced cell-autonomous and paracrine fibrogenic activation, while elevating lipofibroblast fate markers and NAD+ biosynthetic pathways associated with fibrosis resolution.18 These data establish PGC1α as critical to lung fibroblast homeostasis and demonstrate broad beneficial effects of restoring PGC1α in IPF fibroblasts. They also underscore the need to better understand how PGC1α levels are spontaneously elevated during normal resolution of lung injury and remain stably repressed in IPF fibroblasts (figure 1).
One potential mechanism of stable repression of PGC1α is through epigenetic mechanisms. Intriguingly, a recent paper reported that PPARGC1A gene is epigenetically repressed by the DNA methyltransferases 1 (DNMT1) in endothelial cells, and alteration in this transcriptional regulation leads to a dysfunctional mitochondrial biogenesis.37 Additionally, DNMT1 inhibition has been shown to be beneficial in promoting fibrosis resolution in a mouse model of chronic kidney disease38 in which mitochondrial dysfunction is also a disease driver. Future investigation will be needed to further define the relationship between epigenetic regulation, cell metabolism and sustained fibrogenesis in IPF.
Senescence is emerging as an important theme in IPF and other fibrotic disorders,24 39–41 and mitochondrial dysfunction is linked to cellular senescence.42 43 Previous reports from our group and others24 44 45 have highlighted that a subset of activated fibroblasts in IPF and experimental lung fibrosis exhibit a senescent phenotype. Our results demonstrate that PGC1α re-expression in IPF fibroblasts reduces the expression of senescence markers p16 and βGal, consistent with prior evidence of beneficial effects of PGC1α in reducing senescence,46 and strongly influences the secretory profile of these fibroblasts. Our results confirm that IPF fibroblasts secrete abundant soluble mediators belonging to the SASP,47 48 and CM from IPF fibroblasts strongly influences the fibrogenic and senescent state of normal lung fibroblasts. Remarkably, PGC1α re-expression in IPF fibroblasts reduced profibrotic and prosenescence signalling to normal fibroblasts. Additional work will be required to understand the contributions of individual secreted factors to these paracrine effects. Nevertheless, our work supports the concept that IPF fibroblasts, through their secretome, influence the fibrogenic and senescent state of naive bystander cells and that PGC1α re-expression in IPF fibroblasts beneficially alters this secretory profile.
Based on our findings, there is a clear need for further investigation aimed at understanding the mechanisms by which PGC1α reduces profibrotic fibroblast activation. Our results suggest that restoring PGC1α levels improves mitochondrial biogenesis and function, NAD biosynthesis and also promotes the lipogenic fibroblast fate through the induction of PPARγ transcription. Additional efforts will be needed to clarify the inter-relationship of these effects and their functional effects on reducing the activated fibrogenic and secretory state of fibroblasts. One limitation of our study is the relatively small sample size of our in vitro investigations. Further work to define the individual variations in PGC1α expression and function, particularly in IPF-derived cells, will be essential.
An important implication of our work is the need to identify therapeutic strategies that increase PGC1α expression and function in IPF fibroblasts. While recent work has demonstrated a beneficial effect of thyroid hormone in augmenting mitochondrial function and recovery from lung fibrosis in young mice,6 our data demonstrate that T3 hormone is not effective in enhancing PGC1α expression in IPF fibroblasts. In contrast, PGC1α expression was enhanced by the PPARγ agonist rosiglitazone. These results emphasise the need to identify therapeutic strategies effective in disease-derived cells and strongly suggest that cell-specific mechanisms may be required to target PGC1α and mitochondrial function within the lung. Given the broad relevance of mitochondrial function and PGC1α to human physiology, identification of cell and context-specific avenues for therapeutic intervention should be prioritised.
In conclusion, our work implicates PGC1α as an important regulator of fibroblast function and identifies its repression in IPF fibroblasts as key element supporting the contractile, matrix synthetic and secretory phenotype of these cells. Further discovery and development of therapeutic strategies to reverse PGC1α repression in IPF may be beneficial in the treatment of lung fibrosis.
References
Footnotes
Contributors NC, DJT and GL designed the study. NC, JAM, DLJ, QT, AJH, LJM and KMC performed experiments. NC, JAM and GL analysed the data. The manuscript was drafted by NC, DJT and GL and revised by NC, YSP, DJT and GL. All authors participated in manuscript preparation and provided final approval of the submitted work.
Funding This study was funded by National Heart, Lung, and Blood Institute (HL092961, HL133320, HL142596).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.