Article Text

Abstract
Background The role of interleukin 17 (IL-17) in hypoxic pulmonary hypertension (HPH) remains unclear. This study is designed to explore whether IL-17 is a potential target for HPH treatment.
Methods Clinic samples from the lung tissue and serum were obtained from qualified patients. Western blotting, immunohistochemistry and/or ELISA were used to measure the expression of relevant proteins. HPH models were established in C57BL/6 wild-type (WT) and IL-17 −/− mice and were treated with exogenous recombinant mouse IL-17 (rmIL-17) or an IL-17 neutralising antibody. Assays for cell proliferation, angiogenesis and adhesion were employed to analyse the behaviours of human pulmonary arterial endothelial cells (HPAECs). A non-contact Transwell coculture model was used to evaluate intercellular interactions.
Results Expression of IL-17 was increased in lung tissue of both patients with bronchiectasis/COPD-associated PH and HPH mouse model. Compared with WT mice, IL-17 −/− mice had attenuated HPH, whereas administration of rmIL-17 aggravated HPH. In vitro, recombinant human IL-17 (rhIL-17) promoted proliferation, angiogenesis and adhesion in HPAECs through upregulation of Wnt3a/β-catenin/CyclinD1 pathway, and siRNA-mediated knockdown of β-catenin almost completely reversed this IL-17-mediated phenomena. IL-17 promoted the proliferation but not the migration of human pulmonary arterial smooth muscle cells (HPASMCs) cocultured with HPAECs under both normoxia and hypoxia, but IL-17 had no direct effect on proliferation and migration of HPASMCs. Blockade of IL-17 with a neutralising antibody attenuated HPH in WT mice.
Conclusions IL-17 contributes to the pathogenesis of HPH through upregulation of β-catenin expression. Targeting IL-17 might provide potential benefits for alternative therapeutic strategies for HPH.
- hypoxia-induced pulmonary hypertension
- IL-17
- β-catenin
Statistics from Altmetric.com
Key messages
What is the key question?
Whether does interleukin 17 (IL-17) contribute to hypoxic pulmonary hypertension (HPH) and what is the possible mechanism?
What is the bottom line?
IL-17 is involved in HPH, while targeting IL-17 ameliorates pulmonary artery remodelling in HPH.
Why read on?
This is the first report that explores relationships between IL-17/beta-catenin and pulmonary artery remodelling during HPH, identifying a novel target for therapeutics in HPH.
Background
Hypoxic pulmonary hypertension (HPH) is a life-threatening pathophysiological disorder that commonly occurs in patients with chronic lung diseases.1 Most cases of HPH are mild to moderate, but some (approximately1%−4%) patients with HPH suffer severe pulmonary hypertension (PH) with a grim prognosis.2 HPH in chronic lung diseases is associated with increased morbidity and mortality because there are no effective pharmacological treatments for this condition.
The main pathophysiological characteristics of HPH are hypoxic pulmonary vasoconstriction and hypoxic pulmonary vascular remodelling. The latter contributes to a great extent to the increase in pulmonary arterial pressure and pulmonary vascular resistance. The pathogenesis of hypoxic pulmonary vascular remodelling includes chronic inflammation, phenotypic transition, impaired angiogenesis, metabolic shift, apoptosis resistance, pulmonary arterial smooth muscle cell (PASMC) and pulmonary vascular adventitial fibroblast proliferation and pulmonary arterial endothelial cell (PAEC) dysfunction.3
The existing targeted therapies (prostacyclin analogues, endothelin receptor antagonists and phosphodiesterase type 5 inhibitors) lack the ability to reverse the pulmonary vascular remodelling that occurs, and the efficacies of these treatments for HPH are still uncertain.4 Therefore, exploring novel mechanisms underlying pulmonary vascular remodelling during HPH and identifying new therapeutic targets are urgently required. Immune dysregulation and local inflammation contribute to increased pulmonary vascular tone and remodelling.5 6 IL-17A (usually known as IL-17), an important proinflammatory cytokine produced by Th17 cells and other cell types, plays a role through binding to a heteromeric receptor composed of the IL-17RA and IL-17RC subunits and activating the canonical nuclear factor-kappa B, mitogen-activated protein kinase kinase/extracellular signal-regulated kinases1/2, phosphoinositide 3-kinases/protein kinase B, c-Jun N-terminal kinase, and p38 mitogen-activated proteinkinase (NF-κB, MEK-ERK1/2, PI3K-Akt, JNK and p38 MAPK) pathways.7
IL-17 is involved in the pathogenesis of many diseases, including chronic airway inflammatory disease and pulmonary fibrosis, but IL-17-mediated effects during HPH development and their underlying mechanisms are still unclear. In the present study, we proposed to study the role of IL-17 in the pathogenesis of HPH in vitro and in vivo, aiming to find a new target for HPH treatment.
Materials and methods
See the online supplementary file 1 for a more detailed version of these methods.
Supplementary file 1
Subjects
Written informed consent was obtained from all subjects participated. Systolic pulmonary artery pressure of patients with PH was estimated by echocardiography (the details are described in supplements).8 The lung tissues were obtained via lung transplantation or lobectomy both from patients and donors, fixed in the 10% formaldehyde, then embedded in paraffin. Serum samples were collected from patients with COPD or COPD-induced PH (COPD-PH) and control subjects and stored at −80°C for ELISA analysis. The clinic demographic characteristics of these subjects are shown in tables 1 and 2.
Demographic information of bronchiectasis patients providing lung tissues
Demographic information of patients with COPD providing serums
Cell culture
Human PAECs and PASMCs (ScienCell Research Laboratories, California, USA) were cultured in the relevant mediums according to the manufactures’ instruction and maintained at 37°C in a humidified normoxia condition (21% O2, 5% CO2 and 74 % N2). The details are described in supplements.
Cell coculture assay
A non-contact transwell coculture model9 was used to evaluate the role of PAECs dysfunction on proliferation and migration of PASMCs. The details are in supplements.
Cell viability and proliferation assays
Methyl thiazolyl tetrazolium (MTT) and Bromodeoxyuridine (BrdU) incorporation assays were used for analysing cell viability and proliferation. The details are described in supplements.
Tube formation assay
To detect the human PAECs tube formation ability, a tube formation assay was performed. The details are summarised in supplements.
Adhesion assay
Differently treated cells were trypsinised and seeded on top of fibronectin-precoated (30 ul of 20 mg/mL fibronectin per well; BD Bioscience) 96-well plate (1×104/per well in 5% fetal bovine serum (FBS) and growth factor-free endothelial cell medium (ECM)) and incubated for 30 min at 37°C. After washed with phosphate buffer saline (PBS), the adhesion cells were photographed (six fields/per well/each group), and the results were analysed by ImageJ software.10
siRNA preparation and treatment
β-catenin-specific siRNA oligonucleotides were purchased from GenePharma (GenePharma, Suzhou, China). Human PAECs were transiently transfected with siRNA in Opti-MEM medium (Invitrogen, Carlsbad, California, USA) using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer’s recommendation.
Animal models
The study was approved by the Institutional Animal Care and Use Committee of Capital Medical University and followed the ARRIVE Guidelines.11 Murine models of HPH were established as previously described.12 Some of the mice were treated with either recombinant mouse IL-17 (rmIL-17) or a neutralising antibody against mouse IL-17 during the modelling process (the details seen in supplements).
Haemodynamic and ventricular weight measurements
Right ventricular systolic pressure (RVSP) was measured as previously described (the details seen in supplements).12 The weights of the right ventricle (RV) and left ventricle (LV) plus interventricular septum (S) were measured to evaluate the extent of hypertrophy of RV. The right ventricular hypertrophy index (RVHI) was calculated by the formula: RVHI (%)=[RV/(LV +S)]×100.
Lung morphometric analysis
Pulmonary arteriolar remodelling was estimated by per cent media thickness (% MT), % MT=(circumferenceext/π-circumferenceintint/π)/(circumferenceext/π)×100,13 in which circumferenceext and circumferenceint mean the circumferences bounded by the external and internal elastic lamina, respectively (more details seen in supplements).
Immunohistochemical analysis
Immmunoreactivity of IL-17 was detected in paraffin-embedded human lung sections using a rabbit anti-human IL-17 antibody (1:50 dilution, Abcam, UK). Positive signals were visualised using HRP-conjugated goat anti-rabbit secondary antibody (1:200 dilution). The positive cells were developed by diaminobenzidine reagent, and nucleus was stained with haematoxylin.
Western blot analysis
Western blot was used to measure the expression of IL-17, β-catenin, Wnt3a, CyclinD1, β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (the details stated in supplements).
ELISA
The concentrations of IL-17 in serum of human and mice were measured using ELISA kits (the details stated in supplements).
Statistical analysis
Data were presented as means ± SD or Median (IQR). In comparisons with more than two conditions, we have used one way or two way ANOVA followed by post hoc Bonferroni tests for parametric statistical evaluation, and used Kruskal–Wallis test first for nonparametric statistical evaluation followed by the Dunn’s post-hoc test for inter-group comparisons (further details stated in the online supplement). A p value < 0.05 was considered statistically significant. The details of the sample size calculation are stated in the online supplement (online supplementary file 1).
Results
IL-17 expression was upregulated in both patients with bronchiectasis/COPD-associated PH and HPH model mice
Immunohistochemistry showed that the immunoreactivity of IL-17 significantly increased in the lung tissue from subjects with bronchiectasis-induced PH compared with those from patients with bronchiectasis only and those from normal donors (n=3 for each). IL-17-positive signals were predominantly found in the innermost medial layer of the pulmonary arterioles (figure 1A, a, b, c). In addition, the number of IL-17-positive inflammatory cells also increased in lung sections from the group with bronchiectasis-induced PH compared with those from the other groups (figure 1A, d, e, f).

Increased expression of IL-17 in bronchiectasis/COPD-associated PH patients and mouse model. (A) Immunoreactivity for IL-17 (brown) in lung tissues from patients of bronchiectasis with or without PH and a normal control. (a–c) pulmonary arterioles of lung tissues. Immunoreactivity for IL-17 was located predominantly in the innermost medial layer of pulmonary arterioles. The arrows show pulmonary arterioles. Scale bars: 50 µm. (d–f) IL-17-positive cells infiltrated in lung tissues. The arrows show IL-17-positive cells. Scale bars: 25 µm. (g) Bar charts for IL-17 staining of lung tissue sections from patients (three patients for each group). The results are shown as median (IQR), p exact values were obtained following Kruskal-Wallis test. ***P<0.0001 (vs control), ###p<0.0001 (vs bronchiectasis). (B) Concentrations of IL-17 in the serum of patients with COPD with or without PH and normal controls measured by ELISA (n=16 for control group, n=29 for COPD group, n=36 for COPD-PH group). The results are shown as median (IQR), p exact values were obtained following Kruskal-Wallis test. **P=0.0015 (vs control); ###p<0.0002 (vs COPD). (C) Expression of IL-17 in mice exposed to normoxia for 4 weeks (N4W) or to hypoxia for 4 weeks (H4W). (a and b) Scatter plot graphs of RVSP and RVHI in murine models of H4W compared with N4W (n=8 mice for each group). ***P<0.0001. (c–f) Immunoreactivity for IL-17 (brown) in lung tissues of mice from N4W and H4W group. The arrows show pulmonary arterioles, and the black triangles show IL-17-positive cells infiltrated in lung tissues. (c and d) Scale bars: 50 µm; (e and f) Scale bars: 25 µm. (g) Bar charts for IL-17 staining of lung tissue sections from mice. ***P=0.0002. The results are shown as mean±SD, analysed by Student’s t-test. (h) Western blot analysis of IL-17 in lung tissue of murine models. **P=0.0025. The results are shown as mean±SD, analysed by Student’s t-test. (D) The expression of IL-17 in mice exposed to hypoxia for 0 week, 1 week, 2 weeks, 3 weeks and 4 weeks, respectively, and we compared 1 week, 2 weeks, 3 weeks and 4 weeks group with 0 week group, respectively. (a) RVSP and RVHI in mice under hypoxia at different time points (n=8 mice for each group).RVSP: *p=0.0224 (0 week vs 2 weeks), **p=0.0015 (0 week vs 3 weeks), ***p<0.0001 (0 week vs 4 weeks); RVHI: ***p<0.0001 (vs 0 week); the results are shown as mean±SD, analysed by Student’s t-test. (b) Western blot analysis of IL-17 in lung tissue of mice at different time points. *P=0.0286. (c) Concentrations of IL-17 in the serum of mice at different time points. **P=0.0079 (0 week vs 4 weeks). The results are shown as Median (IQR), analysed by Mann-Whitney test. RVSP, right ventricular systolic pressure; RVHI, right ventricular hypertrophy index. IL-17, interleukin 17; PH, pulmonary hypertension.
Retinoic acid-related orphan receptor γt (RORγt), a specific transcription factor for Th17 cells, is necessary for Th17 cell development. The results of an immunohistochemical analysis showed that RORγt expression significantly increased in lung the tissue from subjects with bronchiectasis-induced PH compared with those from patients with bronchiectasis only and those from normal donors (online supplementary figure 1).
An ELISA analysis showed that the concentrations of IL-17 were also higher in the serum from COPD patients with PH compared with that from COPD patients without PH and healthy control subjects (figure 1B).
RVSP (figure 1C, a) and RVHI (figure 1C, b) levels in mice with hypoxia-induced PH (exposed to hypoxia for 4 weeks [H4W]) were significantly elevated compared with those in mice exposed to normoxia for 4 weeks (N4W). Immunohistochemistry showed that the immunoreactivity of IL-17 was significantly increased in the lung sections from mice in the H4W group, IL-17 positive signals were distributed along the endothelium of the pulmonary artery (figure 1C, c, d) and the number of IL-17-positive inflammatory cells that infiltrated the lungs was significantly increased in the H4W group (figure 1C, e, f). Western blot analysis showed that IL-17 expression was also significantly upregulated in the lung tissue homogenate from mice in the H4W group compared with that from the N4W group (figure 1C, h).
We next exposed mice to hypoxia for 0, 1, 2, 3 or 4 weeks. The results revealed that both RVSP and RVHI levels (figure 1D, a) were gradually increased in a time-dependent manner after hypoxia exposure, corresponding to gradual increases in the expression of IL-17 in the lung tissue homogenate and the concentration of serum IL-17 measured by western blotting and ELISA, respectively (figure 1D, b, c). These results suggest that IL-17 may participate in the pathogenesis of HPH.
Knockout of IL-17 attenuated the HPH index in mice undergoing hypoxia exposure
To further understand the influence of IL-17 on hypoxia-induced PH, changes in haemodynamics and pulmonary vascular remodelling were compared between IL-17−/− and wild-type (WT) mice exposed to normobaric N4W or H4W, respectively. The results showed that the body weight gain of IL-17−/− mice was more obvious than that of WT mice under hypoxia (figure 2A), while RVSP significantly increased in H4W WT mice, but the increase was not as strong in H4W IL-17−/− mice (figure 2B). These results indicate that the deletion of the IL-17 gene attenuates hypoxia-induced PH. Because vascular remodelling during PH is mainly seen in the distal pulmonary arteriole, we analysed the % MT of <100 µm intrapulmonary arterioles. The results showed that hypoxia markedly induced <100 µm intrapulmonary arteriole remodelling in WT mice, but not in IL-17−/− mice, especially in % MT of <75 µm pulmonary arterioles (figure 2C). Pulmonary vascular remodelling leads to increase in pulmonary arterial pressure and in right ventricular hypertrophy.12 Therefore, we evaluated the right ventricle of the mice. The RVHI and RV/body weight ratio, two of the indexes of right ventricular hypertrophy, were significantly increased in the WT mice in the H4W group, while these changes were not significant in the IL-17−/− mice (figure 2D).
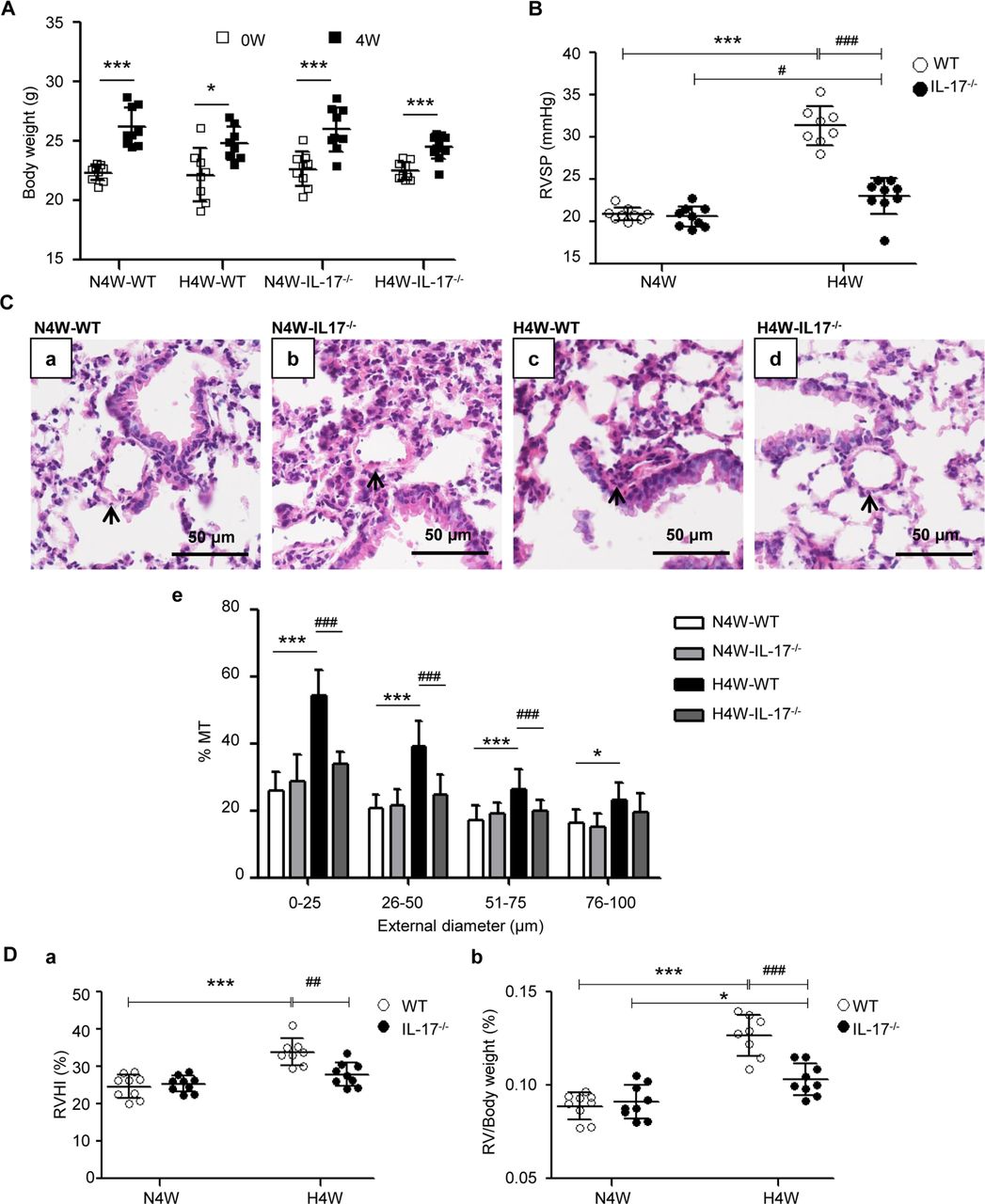
Deletion of the IL-17 gene prevents hypoxia-induced PH. (A) Weight gain of wild-type (WT) and IL-17 knockout (IL-17−/− ) mice exposed to normoxia for 4 weeks (N4W) or hypoxia for 4 weeks (H4W) (n=8–10 mice for each group). *P=0.0121, ***p<0.0001 (vs 0 weeks). The results are shown as mean±SD, analysed by Student’s t-test. H4W- IL-17−/− , IL-17 knockout mice under hypoxia for 4 weeks; H4W-WT, wild-type mice under hypoxia for 4 weeks; N4W-IL-17−/− , IL-17 knockout mice under N4W; N4W-WT, wild-type mice under N4W. (B) RVSP of WT and IL-17−/− mice under normoxia or hypoxia condition for 4 weeks. #P=0.0335, ***p<0.0001, ### p<0.0001. The results are shown as mean±SD, analysed by two-way analysis of variance (ANOVA). (C) Comparison of pulmonary vascular remodelling in WT and IL-17−/− mice under conditions of normoxia or hypoxia for 4 weeks. (a–d) H&E staining of pulmonary arterioles. The arrows show pulmonary arterioles. Scale bars: 50 µm. (e) Percentage medial thicknesses (% MT) of pulmonary arterioles categorised by external diameter into four groups (0–25 µm, 26–50 µm, 51–75 µm and 76–100 µm). 0–25 µm, 26–50 µm and 51–75 µm group: ***p<0.0001, ###p<0.0001; 76–100 µm group: *p=0.0107. Within four groups of pulmonary arterioles, the results are shown as mean±SD, analysed by two-way ANOVA. (D) RVHI and RV/body weight ratio of WT and IL-17 knockout mice exposed to normoxia compared with hypoxia. RVHI: ***p<0.0001, ##p=0.0019; RV/Body weight ratio: *p=0.0482, ***p<0.0001, ###p<0.0001. The results are shown as mean±SD, analysed by two-way ANOVA. IL-17, interleukin 17; PH, pulmonary hypertension;RV, right ventricle; RVHI, right ventricular hypertrophy index; RVSP, right ventricular systolic pressure.
Exogenous IL-17 aggravated HPH
Exogenous rmIL-17 did not affect the weight gain, RVSP level and % MT of the pulmonary arteriole under normoxia (online supplementary figure 2).
We further investigated whether exogenous IL-17 is able to aggravate HPH in mice. rmIL-17 was administered to C57BL/6 mice via intraperitoneal injection at the indicated timepoints during hypoxia exposure, and results were compared with those of mice from the N4W or H4W groups that were not given rmIL-17. As predicted, the body weight was significantly higher in mice from the N4W and H4W groups, but there was no obvious change in mice from the H4W+rmIL-17 group compared with the respective controls (figure 3A).
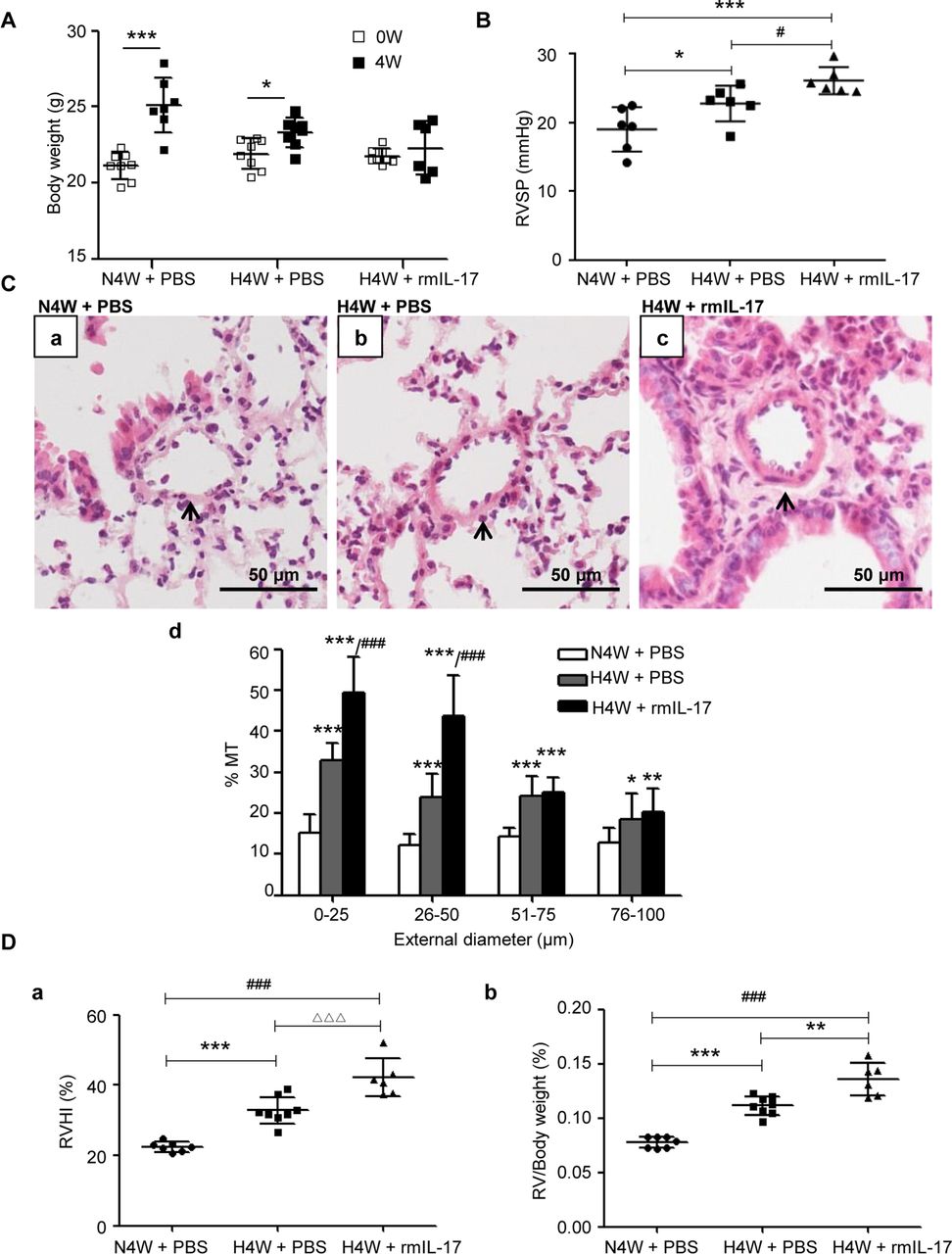
IL-17 aggravates hypoxic pulmonary vascular remodelling. (A) Weight gain of mice exposed to normoxia (N4W+PBS) and hypoxia with (H4W+rmIL-17) or without IL-17 (H4W+PBS) for 4 weeks. n=8 mice for each group, after 4 weeks, one mouse dead in N4W group and two mice dead in H4W+rmIL-17 group. *P=0.0125, ***p=<0.0001, compared with their own basic weight. The results are shown as mean±SD, analysed by Student’s t-test. (B) RVSP of mice exposed to normoxia (N4W+PBS) and hypoxia with (H4W+rmIL-17) or without IL-17 (H4W+PBS) for 4 weeks. RVSP of six mice for each group were successfully detected. *P=0.0262, #p=0.0467, ***p=0.0003. The results are shown as mean±SD, analysed by one-way analysis of variance (ANOVA). (C) Comparison of pulmonary vascular remodelling among groups of N4W+PBS, H4W+PBS and H4W+rmIL-17. (a–c) H&E staining of pulmonary arterioles. The arrows show pulmonary arterioles. Scale bars: 50 µm. (d) % MT of pulmonary arterioles of mice grouped by external diameter. 0–25 µm: ***p<0.0001 (vs N4W+PBS), ###p<0.0001 (vs H4W+PBS); 26–50 µm: ***p<0.0009 (H4W+PBS vs N4W+PBS), ***p<0.0001 (H4W+rmIL-17 vs N4W+PBS), ###p<0.0001 (H4W+rmIL-17 vs H4W+PBS); 51–75 µm and 76–100 µm: *p=0.0481, **p=0.0075, ***p<0.0001 compared with N4W+PBS group. The results are shown as mean±SD, analysed by one-way ANOVA. (D) Right ventricular hypertrophy of mice in the groups of N4W+PBS, H4W+PBS and H4W+rmIL-17. (a) RVHI of mice. RVHI: ***p=0.0002, ###p<0.0001, △△△ p=0.0007. (b) RV/body weight ratio of mice. **P=0.001, ***p<0.0001, ###p<0.0001. The results are shown as mean±SD, analysed by one-way ANOVA. % MT, per cent media thickness; IL-17, interleukin 17; RVHI, right ventricular hypertrophy index; RVSP, right ventricular systolic pressure.
Compared with the control H4W groups, the H4W group treated with exogenous IL-17 exhibited further aggravated HPH, as indicated by increases in RVSP (figure 3B), % MT of <50 µm intrapulmonary arterioles (figure 3C) and right ventricular hypertrophy with elevated RVHI and RV/body weight ratio (figure 3D).
IL-17-mediated dysfunction of human PAECs
It is known that PAECs dysfunction is an important initiating factor for the pulmonary vascular remodelling. Our data showed that the immunoreactivity of IL-17 was located predominantly in the innermost medial layer of the pulmonary arterioles, so we investigated whether IL-17 affects the functions of PAECs. An MTT analysis showed that the maximal proliferation response by PAECs occurred with10 ng/mL IL-17 (figure 4A), which we chose for subsequent experiments as the stimulating concentration, as it was consistent with a previous study.14 Correspondingly, a BrdU incorporation assay showed that 10 ng/mL rhIL-17 increased the percentage of BrdU-stained PAECs (P4, S phase of the cell cycle), indicating IL-17-induced the proliferation of PAECs (figure 4B). In tube formation assays, recombinant human IL-17 (rhIL-17) significantly increased the tube length of the formed tubes (figure 4C), suggesting enhanced angiogenic abilities in PAECs in vitro. In addition, rhIL-17 also increased the adhesive capacity of PAECs (figure 4D). Together, these findings suggest that rhIL-17 is able to promote endothelial cell proliferation, angiogenesis and adhesion in vitro.

Effects of exogenous IL-17 on human pulmonary arterial endothelial cells (PAECs). (A) Effects of IL-17 on the proliferation of PAECs measured by MTT. Base versus 10 ng/mL: ***p<0.0001; 0 ng/mL versus 10 ng/mL: **p=0.0058; 10 ng/mL versus 20 ng/mL: **p=0.006; 10 ng/mL versus 50 ng/mL: ***p=0.0002. The results are shown as mean±SD, analysed by one-way ANOVA. (B) Effects of IL-17 on the cell cycle positions and DNA synthetic activities of PAECs determined by analysing total DNA and incorporated BrdU using flow cytometry. The cells were treated with or without IL-17 (10 ng/mL) for 24 hours. Gate P3 represents cells in G0/G1 phase, P4 is S phase, P5 is G2+M phase and P6 is apoptotic cells. **P=0.0036. (C) Spontaneous formation of capillaries by PAECs cultured with or without IL-17 (10 ng/mL) for 24 hours. The result is expressed as the mean total tube lengths per field, data are calculated from at least 6 fields of view for every replicate and at least three replicates per group are set for every experiment. Scale bars: 500 µm. **P=0.0012. (D) Adhesion of PAECs to culture plate wells when treated with or without IL-17 (10 ng/mL) for 24 hours. Adherent cells were counted and expressed as the mean numbers of cells per high power field, data are calculated from at least six fields of view for every replicate and at least three replicates per group are set for every experiment. Scale bars: 200 µm. ***P=0.0009. The results are shown as mean±SD, analysed by Student’s t-test. ANOVA, analysis of variance; IL-17, interleukin 17; MTT, methyl thiazolyl tetrazolium.
Dysfunction of human PAECs induced by IL-17 affected the proliferation but not the migration of human PASMCs
Because interactions between PAECs and PASMCs contribute to pulmonary vascular remodelling, we investigated the effects of PAECs dysfunction on PASMCs using a non-contact Transwell coculture model in the presence/absence of rhIL-17. As shown in figure 5A, the proliferation of PASMCs was significantly promoted in the presence of rhIL-17 when PASMCs were cocultured with PAECs. However, the same model did not affect the migration of PASMCs (figure 5B).
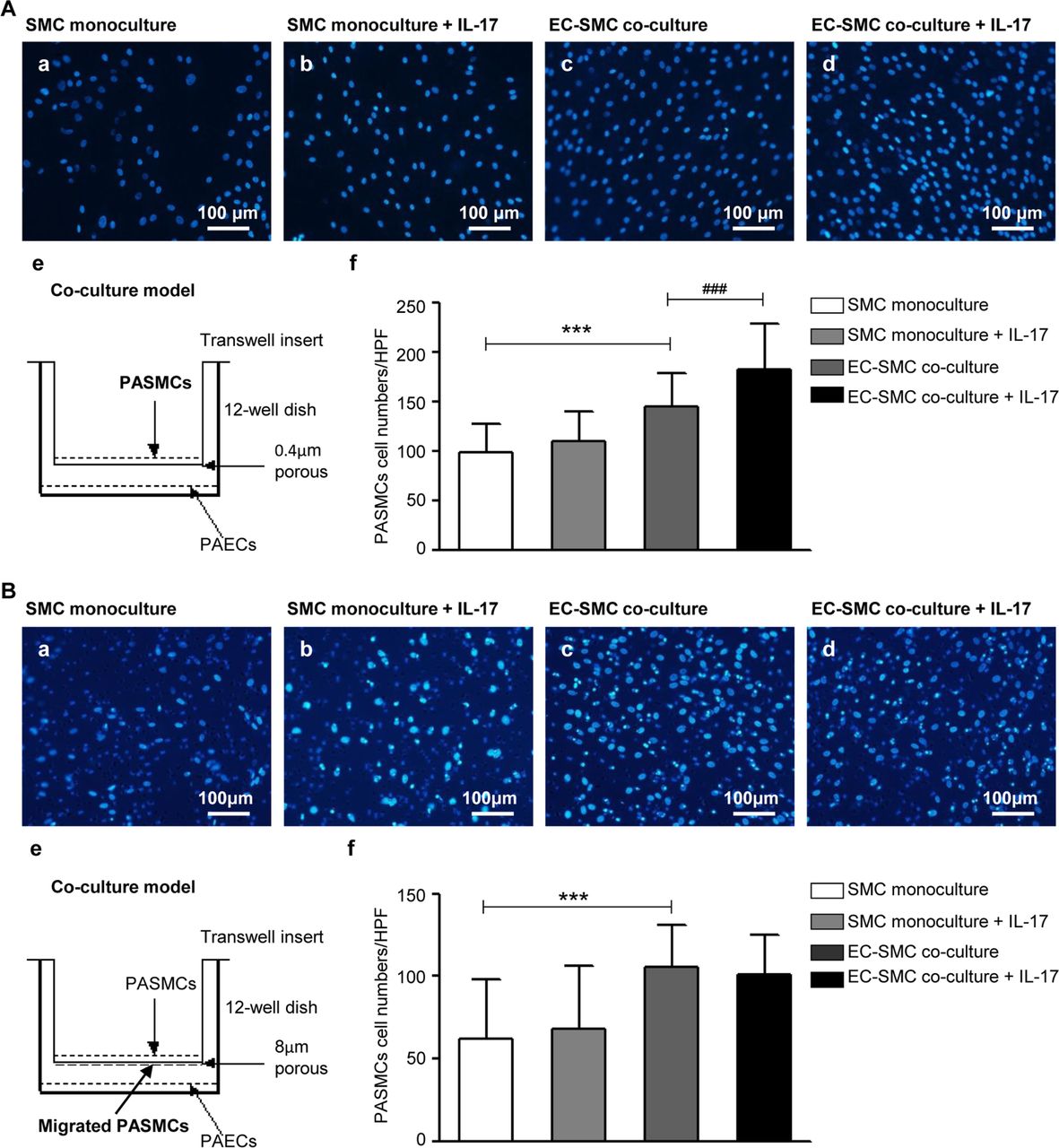
Effects of IL-17 induced human PAECs dysfunction on human PASMCs. (A) IL-17 induced PAECs dysfunction promotes PASMCs proliferation. (a–d) The nuclei of PASMCs were stained with DAPI. Scale bars: 100 µm. (e) Schematic of the Transwell coculture model. PASMCs were seeded in the top compartment, separated by a porous membrane from PAECs that were cultured in the bottom compartment. In this system, the pore size of porous membrane is 0.4 µm, which cannot allow cell migration. (f) Quantification of proliferating cells. Data are representative of mean numbers of cells per high power field (HPF), at least six fields of view for every replicate are calculated, and at least three replicates per group are set for every experiment. ***P<0.0001, ###p=0.0001. (B) IL-17-induced PAECs dysfunction has no effects on PASMCs migration. (a–d) The nuclei of PASMCs were stained with DAPI. (e) Schematic of the Transwell coculture model. In this system, the pore size of porous membrane is 8 µm, which allows cell migration. (f) Quantification of migrating cells. Data are representative of mean numbers of cells per HPF, at least six fields of view for every replicate are calculated and at least three replicates per group are set for every experiment. ***P<0.0001. The results are shown as mean±SD, analysed by two-way ANOVA. SMC monoculture: only PASMC was cultured in the model; SMC monoculture+IL-17: only PASMC was cultured in the model and stimulated by IL-17; EC-SMC coculture: PAEC and PASMC were cocultured in the model; EC-SMC coculture+IL-17: PAEC and PASMC were cocultured in the model and stimulated by IL-17. ANOVA, analysis of variance; EC-SMC, endothelial cell-smooth muscle cell; IL-17, interleukin 17; PAECs, pulmonary arterial endothelial cells; PASMCs, pulmonary arterial smooth muscle cells; SMC, smooth muscle cell.
We then exposed the EC-SMC coculture with and without IL-17 treatment to hypoxia or normoxia. The results showed that IL-17 promoted the proliferation of PASMCs under both normoxia and hypoxia when PASMCs were cocultured with HPAECs. The effect of IL-17 under normoxia and hypoxia is similar (figure 6A). However, IL-17 did not regulate PASMCs migration when PASMCs were cocultured with PAECs under either normoxia or hypoxia (figure 6B).
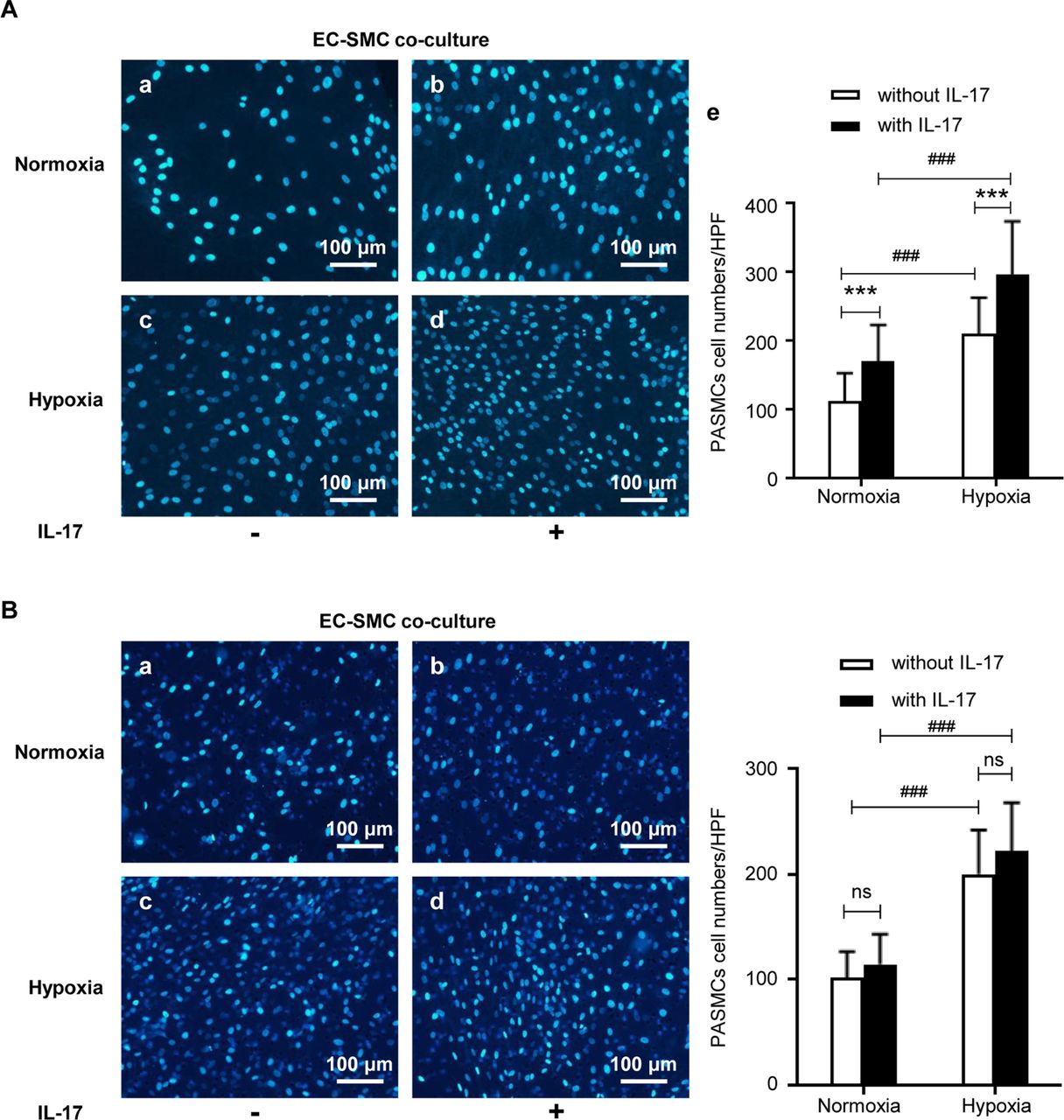
Effects of IL-17 on human PASMCs when cocultured with human PAECs under hypoxia or not. (A) In EC-SMC coculture model, IL-17 promoted the proliferation of PASMCs under both normoxia and hypoxia. (a–d) The nuclei of PASMCs were stained with 4',6-diamidino-2-phenylindole (DAPI). Scale bars: 100 µm. (e) Quantification of proliferating cells. ***P<0.0001, ### p<0.0001. (B) In EC-SMC coculture model, IL-17 had no effect on the migration of PASMCs under both normoxia and hypoxia. (a–d) The nuclei of PASMCs were stained with DAPI. Scale bars: 100 µm. (e) Quantification of migrating cells. ###P<0.0001. The results are shown as mean±SD, analysed by two-way analysis of variance. HPF, high power field; IL-17, interleukin 17; ns, no significance; PAECs, pulmonary arterial endothelial cells; PASMCs, pulmonary arterial smooth muscle cells.
Blockade of IL-17 attenuated HPH in mice
To investigate whether blocking IL-17 can prevent HPH, a neutralising monoclonal antibody against mouse IL-17 was intraperitoneally administered to HPH mice. Compared with control treatment, the blockade of IL-17 significantly reduced the hypoxia-induced increase in RVSP (figure 7B) and <100 µm intrapulmonary arteriole remodelling (figure 7C), although there were no obvious changes in the body weight, RVHI and RV/body weight ratio (figure 7A,D).
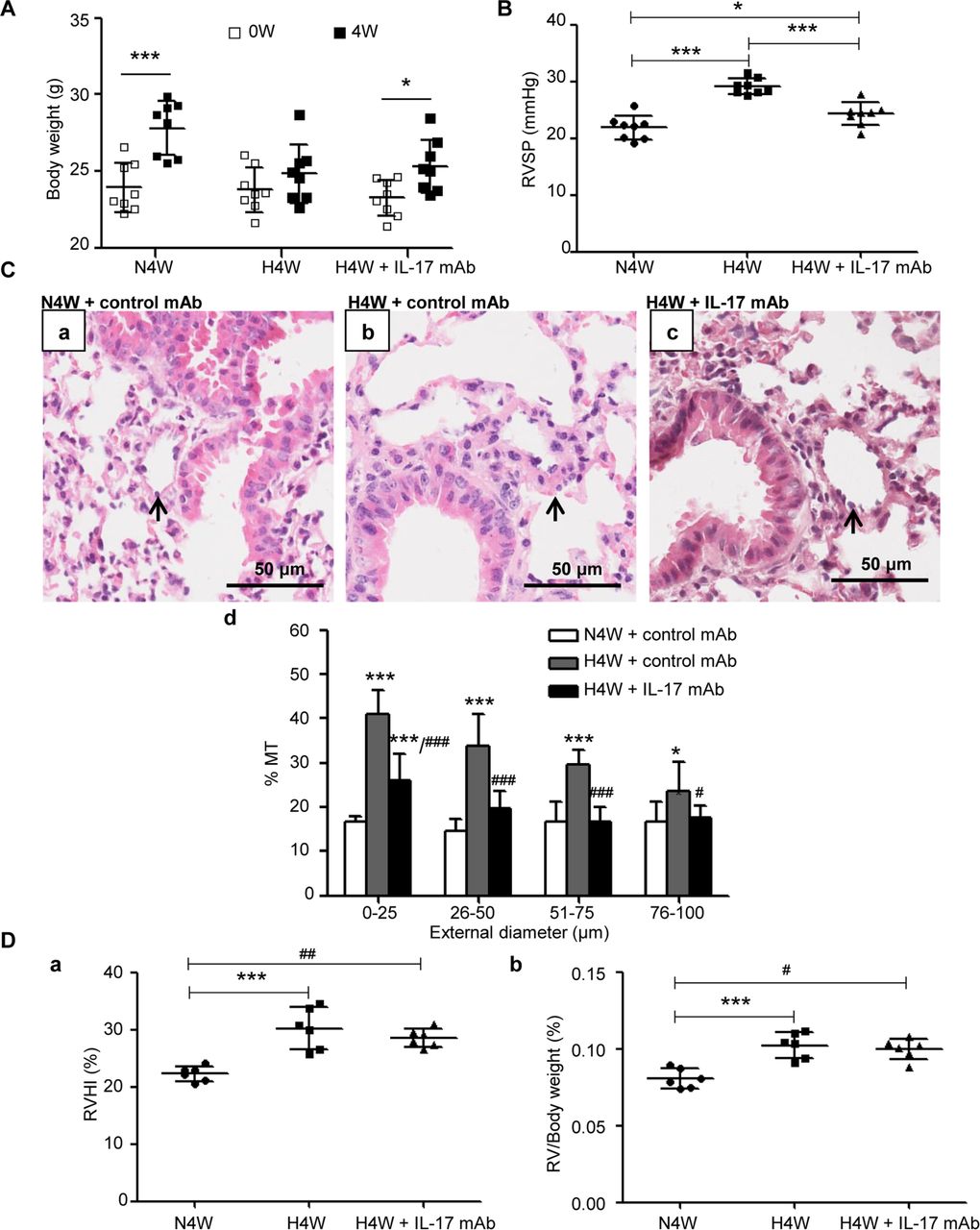
Neutralisation of IL-17 by monoclonal antibody is protective against HPH. (A) Weight gain of mice exposed to normoxia and hypoxia with or without monoclonal antibody against mouse IL-17 (IL-17 mAb) (n=8 mice for each group). *P=0.0142, ***p=0.0004 compared with their own basic weight. The results are shown as mean±SD, analysed by Student’s t-test. (B) RVSP of mice exposed to normoxia (N4W+control mAb) and hypoxia with (H4W+IL-17 mAb) or without IL-17 (H4W+control mAb) for 4 weeks. *P=0.0387, ***p<0.0001. (C) Comparison of pulmonary vascular remodelling among groups of N4W+control mAb, H4W+control mAb and H4W+IL-17 mAb. (a–c) H&E staining of pulmonary arterioles. The arrows show pulmonary arterioles. Scale bars: 50 µm. (d) % MT of pulmonary arterioles of mice grouped by external diameter. *P=0.0109, ***p<0.0001 (vs N4W+control mAb). #P=0.0309, ### p<0.0001 (vs H4W+control mAb). (D) Right ventricular hypertrophy of the above three groups. Right ventricles of six mice for each group were separated successfully. (a) RVHI of mice. ***P=0.0001, ##p<0.0013. (b) RV/body weight ratio of mice. ***P=0.0008, #p=0.0133. The results are shown as mean±SD, analysed by one-way ANOVA. % MT, per cent media thickness; IL-17, interleukin 17; HPH, hypoxic pulmonary hypertension; RV, right ventricule; RVHI, right ventricular hypertrophy index; RVSP, right ventricular systolic pressure.
IL-17 aggravated HPH by upregulating β-catenin
The above results showed that the increased expression of IL-17 played a role in pulmonary vascular remodelling both in vitro and in vivo, so we then explored the potential mechanism. Several studies have shown that IL-17 signalling pathways could increase the activity of Wnt mediators,15–17 and our previous studies showed that β-catenin is an HPH-related gene.18 19 In the present study, we found that the knockdown of the IL-17 gene downregulated the expression of β-catenin (figure 8A), which is consistent with a report by Hyun and colleagues.20 We then examined the role of β-catenin in mediating IL-17-aggravated hypoxia-induced pulmonary vascular remodelling. As expected, β-catenin expression was upregulated in the murine model of HPH (figure 8B) and was further enhanced in the presence of exogenous rmIL-17 (figure 8B). Correspondingly, the administration of a neutralising antibody against mouse IL-17 completely reversed the hypoxia-induced expression of β-catenin (figure 8C), suggesting the possibility that IL-17 promotes HPH by upregulating the expression of β-catenin. In vitro, rhIL-17 also upregulated the expression of β-catenin in PAECs in a time-dependent manner (figure 8Da) and upregulated Wnt3a and CyclinD1 expression, suggesting that the Wnt3a/β-catenin/CyclinD1 pathway was activated (figure 8Db).
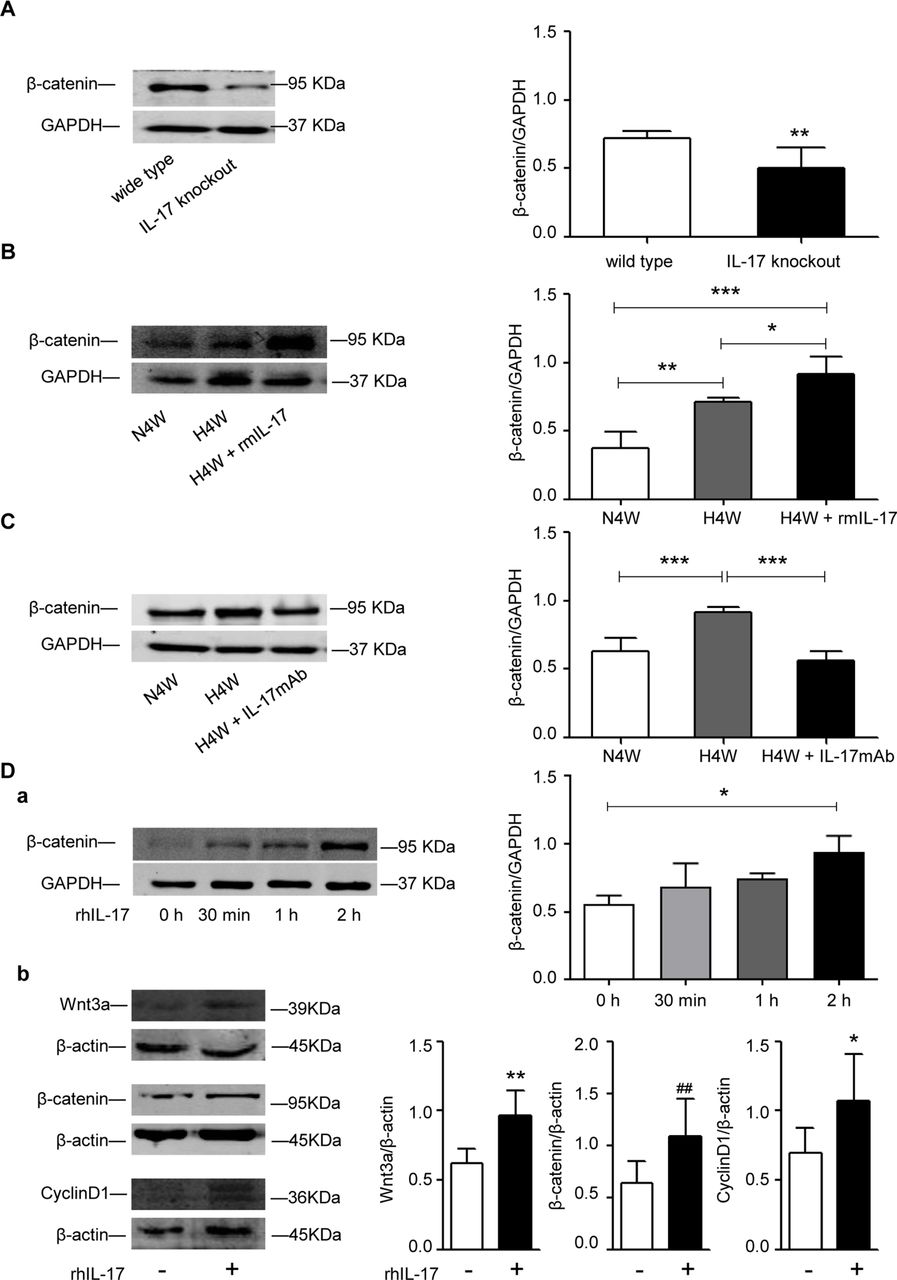
IL-17 regulates the expression of β-catenin. (A) Western blot analysis of β-catenin in lung tissues of WT and IL-17−/− mice. **P=0.0079. The results are shown as mean±SD, analysed by Student’s t-test. (B) Western blot analysis of β-catenin in mice exposed to normoxia and hypoxia with or without rmIL-17. *P=0.0227, **p=0.0014, ***p<0.0001. The results are shown as mean ±SD, analysed by one-way ANOVA. (C) Western blot analysis of β-catenin in mice exposed to normoxia and hypoxia with or without IL-17 mAb. N4W versus H4W: ***p=0.001; H4W versus H4W+IL17 mAb: ***p=0.0002. The results are shown as mean±SD, analysed by one-way ANOVA. (D) The expression of Wnt3a/β-catenin/CyclinD1 pathway in HPAECs induced by rhIL-17. (a) Western blot analysis of β-catenin in HPAECs treated by IL-17 (10 ng/mL) for 0 hour, 30 min, 1 hour and 2 hours respectively. *P=0.0451. The results are shown as median (IQR), p exact values were obtained following Kruskal-Wallis test. (b) Western blot analysis of Wnt3a/β-catenin/CyclinD1 in HPAECs treated by IL-17 for 2 hours. *P=0.0259, **p=0.0060, ##p=0.0091. The results are shown as mean±SD, analysed by Student’s t-test. ANOVA, analysis of variance; IL-17, interleukin 17; H4W, hypoxia for 4 weeks; HPAECs, human pulmonary arterial endothelial cells; mAb, monoclonal antibody; N4W, normoxia for 4 weeks; rhIL-17, recombinant human IL-17; rmIL-17, recombinant mouse IL-17; WT, wild type.
To investigate whether β-catenin mediates IL-17-induced dysfunction in PAECs, we tested the proliferation, angiogenesis and adhesion of cultured PAECs in which β-catenin expression was knocked down using a specific siRNA. The results revealed that the knockdown of β-catenin expression significantly reduced the tube formation by PAECs both in control and IL-17-induced conditions (figure 9A), and similar effects were also observed on IL-17-induced proliferation (figure 9C). For cell adhesion, the knockdown of β-catenin expression did not affect the adhesive capacity of PAECs in the control condition, but it could abrogate IL-17-induced adhesion in PAECs (figure 9B). These results indicated that IL-17 contributes to HPH by upregulating expression of β-catenin.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-17 regulates the functions of HPAECs via β-catenin. (A) Effects of knock down of β-catenin expression with siRNA on IL-17-induced angiogenesis by HPAECs. The result is expressed as the mean total tube lengths per field, data are calculated from at least six fields of view for every replicate and at least three replicates per group are set for every experiment. Scale bars: 500 µm. **P=0.0098, ***p<0.0001, ###p<0.0001. (B) Effects of knockdown of β-catenin expression with siRNA on IL-17-induced adhesion of HPAECs. The results are expressed as mean numbers of cells per high power field, data are calculated from at least six fields of view for every replicate and at least three replicates per group are set for every experiment. Scale bars: 200 µm. ***P<0.0001, ###p<0.0001. (C) Effects of knockdown of β-catenin expression with siRNA on IL-17-induced proliferation of HPAECs. *P=0.0294, ***p=0.0007, ###p<0.0001. The results are shown as mean±SD, analysed by two-way analysis of variance. HPAECs, human pulmonary arterial endothelial cells; IL-17, interleukin 17.
Discussion
It is known that IL-17 is involved in the pathogenesis of many diseases, but its role in HPH is still unclear. In the present study, we demonstrated that: (1) IL-17 expression was upregulated in both patients with bronchiectasis/COPD and elevated pulmonary arterial pressure and a murine model of HPH; (2) the knockout of the IL-17 gene was protective against HPH, while exogenous IL-17 administration aggravated HPH in mice; (3) IL-17 enhanced hypoxia vascular remodelling by mediating dysfunction in PAECs, and PAEC dysfunction promoted proliferation but not migration in PASMCs; (4) neutralising IL-17 attenuated the vascular remodelling induced by hypoxia; and (5) IL-17 participated in HPH through upregulating β-catenin expression. To our knowledge, this is the first report to demonstrate that IL-17 is involved in HPH via β-catenin, suggesting that IL-17 is a potential therapeutic target for HPH.
It is known that dysregulated inflammation and immunity have strong influences on the onset and progression of pulmonary vascular diseases. First, PH is a frequent manifestation of multiple autoimmune conditions.21 For example, the presence of autoantibodies is common in patients with PH.22 23 Second, inflammation is a prominent feature of the pathology of pulmonary vascular remodelling. Increased quantities of inflammatory cells have been found in the perivascular, intravascular areas of vascular remodelling and in plexiform lesions.24 25 Furthermore, altered immune cell populationsand cytokine and chemokine levels have been observed in the blood of patients with PH.26–28 Nevertheless, infectious diseases are often related to PH development.29 In this study, the increased infiltration of IL-17-positive inflammatory and immune cells in the lung tissue was observed, indicating the significant role of inflammation in the pathogenesis of HPH.
Although the serum concentration of IL-17 in patients with COPD-associated PH was markedly elevated compared with that of the control, it was not high as previously reported.30 This low serum level of IL-17 has also been observed by other groups.31
It is well known that IL-17 can act on many cells, including inflammatory cells and structural cells, such as pulmonary endothelial cells, epithelial cells and fibroblasts.32 Although some studies have shown that Th17-related cytokines are upregulated in PH,30 33 34 and IL-17 mainly participates in chronic airway inflammatory disease35 and pulmonary fibrosis,36 there is a lack of information about the role of IL-17 in pulmonary vascular disease. Our data demonstrate that hypoxia upregulates the expression of IL-17 which, as an aggravating factor, mediates pulmonary vascular remodelling by affecting the functions of PAECs in HPH. Liu et al also reported that IL-17 participated in vascular remodelling in transplant vasculopathy by inducing endothelial nitric oxide (NO) synthase expression and NO production inhuman vascular endothelial cells.14
Maston et al 34 found that RAG1 −/− mice, which lack mature T and B cells, exhibited and attenuated HPH phenotype, while the adoptive transfer of CD4+ T cells or Th17 cells could restore this process. However, the role of IL-17 in PH pathology is unknown. We chose IL-17 −/− and WT mice for our HPH model and found that IL-17 −/− mice were resistant to the development of HPH.
PAECs and PASMCs are the most crucial components of the pulmonary vascular wall, and the interactions between them are essential for the maintenance of normal pulmonary vascular physiology and also during the process of pulmonary vascular diseases. Furthermore, pulmonary vascular endothelial dysfunction is an important initiating factor in the development of PH.37 Our data showed that IL-17 significantly enhanced the angiogenesis of PAECs, which is consistent with the observation of Numasaki et al.38 In their study, IL-17 promoted cord formation by vascular endothelial cells in vitro, and tumours transduced with the IL-17 gene had significantly higher vascular density in vivo, indicating that IL-17 mediated disordered angiogenesis. In the study of neoplastic diseases, the data showed that IL-17 promoted tumour growth by enhancing angiogenesis, as greater microvascular density was detected in the recombinant IL-17-treated tumour-bearing mice compared with the non-treated group.39 Although some studies have shown that IL-17 only promotes cell growth in vivo, but not in vitro,38 39 our results revealed that IL-17 was able to induce the proliferation of PAECs at 10 ng/mL, but not at higher concentrations, suggesting that such effect of IL-17 is strictly concentration dependent. A recent study showed that IL-17 (100 ng/mL and 500 ng/mL) could increase the proliferation of human retinal vascular endothelial cells,40 which further supports the proproliferation effect of IL-17 on endothelial cells. For human umbilical vein endothelial cells and pulmonary microvascular endothelial cells, IL-17 has no direct effect on proliferation.38 41 The different results in some reports may be due to the use of different endothelial cells with different vascular origins, or differences in drug concentrations and culture conditions, and the potential mechanisms should be further explored. Because IL-17 could upregulate intercellular cell adhesion molecule 1 expression,40 we examined the adhesion capability of PAECs in response to IL-17 and found that IL-17 stimulated adhesion in PAECs. Furthermore, we examined the effects of IL-17-induced PAECs dysfunction on PASMCs, which was not previously reported. Using a non-contact Transwell EC-SMC coculture model, we found that in the presence of IL-17, cocultured with PAECs promoted the proliferation, but not migration of PASMCs. Taken together, these results suggest that IL-17 causes PAEC dysfunction by directly acting on the PAECs, which affects the function of PASMCs, resulting in the pathogenesis of HPH.
Although a number of signalling pathways are activated by IL-17, such as the canonical NF-κB, MEK-ERK1/2, PI3K-Akt, JNK and p38 MAPK pathways,14 pathways mediated by IL-17 in pathological conditions are poorly elucidated. Our previous studies found that β-catenin is an HPH-related gene.18 19 Here, we found that the deletion of the IL-17 gene downregulated the expression of β-catenin, while exogenous IL-17 upregulated the expression of β-catenin both in vitro and in vivo. Moreover, the knockdown of β-catenin expression blocked the IL-17-mediated dysfunctions of PAECs. All of these results indicate that β-catenin is one of the downstream mediators of an IL-17-elicited signalling pathway. In vivo experiments further revealed that IL-17 could activate the Wnt3a/β-catenin/CyclinD1 pathway in PAECs.
Our data also showed that the blockade of IL-17 with a neutralising antibody could prevent hypoxic pulmonary vascular remodelling, which might provide an alternative therapeutic target for HPH treatment. Currently, the efficacy of three biological agents targeting IL-17 signalling, secukinumab (anti-IL-17A mAb), ixekizumab (anti-IL-17A mAb) and brodalumab (anti-IL-17RA mAb), have been evaluated in the clinic, and the assessments have mainly focused on autoimmune diseases such as psoriasis, rheumatoid arthritis and asthma.42 The side effects of these agents need to be evaluated further.
So far, few studies have explored the role of IL-17 in right ventricular hypertrophy. It has been reported that IL-17 participates in arrhythmogenic RV cardiomyopathy,43 and many lines of evidence support the idea that IL-17 is implicated in left ventricular remodelling.44–47 Our results showed that IL-17 expression in RV tissue was increased during hypoxia (online supplementary figure 3), and exogenous IL-17 promoted IL-17 expression in RV tissue (online supplementary figure 4). The effect of IL-17 on the RV and the potential underlying mechanisms might be elucidated in a future study.
Although the results provided in this work relate IL-17 with patients with bronchiectasis/COPD and elevated pulmonary arterial pressure, it should be noted that human samples used are small, which may limit the interpretation of this work. Another limitation of this work is that the selection of patients with PH for the study was performed by echocardiogram, rather than the gold standard right heart catheterisation examination.
In conclusion, this work implicates IL-17 as a mediator of HPH, and targeting IL-17 is a potential novel anti-inflammatory therapeutic strategy for the clinical treatment of HPH.
Acknowledgments
We would like to thank Professor Hu-ying Shen from Baylor College of Medicine, Professor Ying Sun from Capital Medical University and Dr Bin Liu from Chinese Academy of Medical Sciences and Peking Union Medical College for providing language help and writing assistance.
References
Footnotes
Contributors LW participated in the design of the study, data acquisition, statistical analysis and drafting of the manuscript. JL, WW and XQ participated in acquisition of the data. YW participated in collecting serum samples. BT participated in collecting lung tissues from patients and donor. HD provided the IL-17 knockout mice and participated in the design of the study. JW participated in the design of the study and revised the manuscript. CW, TY and WN conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Funding This paper was supported by National Natural Science Foundation of China (81400039, 91643115 and 81470258).
Disclaimer Funding entities did not contribute to the study design or data collection, analysis and interpretation and the writing of the manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study was approved by the Research Ethics Committee of Capital Medical University and Beijing Chao-Yang Hospital of Capital Medical University and China-Japan Friendship Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.