Article Text

Abstract
Background Despite advances in understanding the pathophysiology of acute respiratory distress syndrome, effective pharmacological interventions have proven elusive. We believe this is a consequence of existing preclinical models being designed primarily to explore biological pathways, rather than predict treatment effects. Here, we describe a mouse model in which both therapeutic intervention and ventilation were superimposed onto existing injury and explored the impact of β-agonist treatment, which is effective in simple models but not clinically.
Methods Mice had lung injury induced by intranasal lipopolysaccharide (LPS), which peaked at 48 hours post-LPS based on clinically relevant parameters including hypoxaemia and impaired mechanics. At this peak of injury, mice were treated intratracheally with either terbutaline or tumour necrosis factor (TNF) receptor 1-targeting domain antibody, and ventilated with moderate tidal volume (20 mL/kg) to induce secondary ventilator-induced lung injury (VILI).
Results Ventilation of LPS-injured mice at 20 mL/kg exacerbated injury compared with low tidal volume (8 mL/kg). While terbutaline attenuated VILI within non-LPS-treated animals, it was ineffective to reduce VILI in pre-injured mice, mimicking its lack of clinical efficacy. In contrast, anti-TNF receptor 1 antibody attenuated secondary VILI within pre-injured lungs, indicating that the model was treatable.
Conclusions We propose adoption of a practical framework like that described here to reduce the number of ultimately ineffective drugs reaching clinical trials. Novel targets should be evaluated alongside interventions which have been previously tested clinically, using models that recapitulate the (lack of) clinical efficacy. Within such a framework, outperforming a failed pharmacologic should be a prerequisite for drugs entering trials.
- ARDS
- critical care
- pulmonary oedema
- innate immunity
Statistics from Altmetric.com
Key messages
What is the key question?
How can we design models of acute lung injury/acute respiratory distress syndrome to have better predictive power, and evaluate them, in a preclinical setting?
What is the bottom line?
A ‘two-hit’ mouse model of acute lung injury, in which therapeutic intervention and ventilation were superimposed onto existing injury, provided better predictions of clinical β-agonist efficacy than when treatment was delivered into healthy lungs in a simple one-hit model.
Why read on?
In our study, we propose a framework whereby knowledge of failed clinical pharmacologics is used to validate new experimental models and targets, to reduce the likelihood of ineffective treatments being tested in patients.
Introduction
Acute respiratory distress syndrome (ARDS) is a frequently fatal condition caused by an overwhelming inflammatory response within the lung to a variety of insults. Despite widespread acceptance that inflammation is intimately linked to pathophysiology, none of the numerous pharmacological targets identified from preclinical studies have translated to patient benefit.
This lack of progress has led to much discussion regarding development of better paradigms in animal studies that may provide closer predictions of patient outcomes. Thus, a host of models have been reported or proposed1–3 utilising systems ranging from rodents to human tissue, varying in complexity from simple ‘single-hit’ models to those involving multiple challenges. Logically, the more closely a model mimics the patient scenario, the more reliable any predictions are likely to be. Practically, the numerous aetiologies of clinical ARDS means that no single model will ever truly capture the situation in all sufferers. However, there are factors common to the majority of patients including (i) mechanical ventilation, an essential common denominator of ARDS treatment (indeed perhaps the main thing patients have in common); (ii) deterioration in oxygenation and respiratory mechanics and (iii) the fact that patients primarily present for treatments only once they are sick. Thus, we would propose that ‘predictive’ models (as opposed to those exploring biological processes) should examine therapeutic delivery into physiologically injured, mechanically ventilated lungs. Therefore, we developed a ‘two-hit’ model in which ARDS-like symptoms were induced in mice by intranasal lipopolysaccharide (LPS), and at the peak of injury intervened therapeutically and superimposed a secondary ventilator-induced lung injury (VILI).
A second, more complicated issue is, having developed any model, how does one evaluate whether it is ‘better’ than existing ones? Given the lack of specific biomarkers in ARDS, there is no single readout one can measure in models that would determine their validity. We have considered an alternative approach to this question. The fact that many biological targets appear effective in animal models but not in patients indicates that existing models are susceptible to producing false-positive results. Therefore, to validate our model, we explored the response to β-agonist treatment which may be considered a clear example of a false positive, effective in various preclinical models,4–7 but ineffective in patients.8 9
We show here that the two-hit model was insensitive to intratracheal β-agonist (terbutaline). This contrasted with the clear beneficial effect of terbutaline within a one-hit pure VILI model, indicating that the response of the two-hit model was closer to that observed in patients. Importantly, we also showed that the model was amenable to intervention by an alternative approach, tumour necrosis factor (TNF) receptor-1-specific blockade. We propose a general framework in which new models and pharmacological targets are evaluated preclinically alongside mediators that have been previously clinically trialled. We feel that such an approach currently represents the best way to validate predictions from preclinical models, and should increase our ability to identify false positives before they enter patient trials.
Methods
Experiments were carried out according to the GSK Policy on the Care, Welfare and Treatment of Animals and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines for the use and reporting of animals in research, under the Animals (Scientific Procedures) Act 1986 UK. In all, 80 male C57BL/6 mice (Charles River) aged 9–12 weeks (25–31 g) were used. Mice were housed in environmentally enriched, individually ventilated cages, maximum five mice/cage. Mice had free access to food and water, were maintained under a 12-hour light:dark cycle and had welfare assessed daily.
LPS-induced lung injury model
Mice were intranasally dosed with 25 µg UltraPure LPS (Escherichia coli O111:B4) under 2% isoflurane. At predetermined time points, respiratory mechanics and arterial blood gases were assessed as described previously.1 In brief, mice were anaesthetised (intraperitoneal ketamine 80 mg/kg, xylazine 8 mg/kg) and an endotracheal tube and carotid artery cannula introduced. Animals were ventilated using a custom-made ventilator10 with 8 mL/kg tidal volume (VT), 3 cm H2O positive end-expiratory pressure (PEEP), using 100% O2. Following a recruitment manoeuvre (30 cm H2O, 5 s) ventilation was continued for 30 min to standardise lung history. Respiratory mechanics were then assessed by end-inflation occlusion and PaO2 determined.
Bronchoalveolar lavage fluid was evaluated for cytokines by ELISA and protein content by Bradford assay. Lung leukocytes were investigated using flow cytometry.11 Lungs were fixed and disrupted using a tissue dissociator. Samples were filtered (40 µm), incubated with fluorescent antibodies against CD45, CD11b, CD11c, Ly6C, Ly6G, F4/80, CD206, ICAM-1 and nitric oxide synthase 2 (NOS2), and analysed using a CyAn flow cytometer with Summit software.
Ventilator-induced lung injury
Two models of VILI were used within the current study, to evaluate the effects of interventions delivered into either healthy or injured lungs. Mice were anaesthetised and instrumented as described above. To model ‘pure’ VILI, healthy mice were ventilated with a high VT of 30–35 mL/kg (PEEP 4 cm H2O, 90 breaths/min, 1:2 inspiratory:expiratory ratio, using 96%O2/4%CO2 to maintain normocapnia). Ventilation was continued for 3 hours (without recruitments) or until mice reached a mortality surrogate (arterial pressure <40 mm Hg). As a two-hit model, 48 hours after LPS (identified as peak injury), mice were ventilated using a moderately high VT of 20 mL/kg (PEEP 4–5 cm H2O, 90 breaths/min, 1:2 inspiratory:expiratory ratio). Inspired gas was 99%O2/1% CO2. Recruitment manoeuvres (30 cm H2O, 5 s) were carried out every 15 min as we have previously shown that derecruitment occurs even with 20 mL/kg in mice,10 and ventilation was continued for 3 hours or until mortality surrogate was reached.
We additionally incorporated various control experiments to clarify that the model was truly two hit in nature, that is, that both insults (LPS pre-injury and moderate VT ventilation) were required to produce substantial physiological injury. First, a group of 48 hours LPS-injured animals were ventilated with low VT (8 mL/kg, PEEP 4 cm H2O, 100% O2). Second, healthy uninjured mice were ventilated with the moderate 20 mL/kg VT. Both control groups were ventilated for 3 hours with recruitment manoeuvres every 15 min.
Interventions
Mice within both the one-hit and two-hit VILI strategies were randomly allocated to receive 50 µL of saline or 10−4M terbutaline intratracheally via the endotracheal tube. This was delivered immediately before the onset of high or moderate VT ventilation, with recruitment manoeuvres (4×30 cm H2O, 5 s) to aid distribution. In addition, mice within the two-hit model only were randomly allocated to receive 100 µg TNF receptor 1-targeting domain antibody (dAb; Dom-1m-15–12; Biopharmaceuticals R&D, GlaxoSmithKline, Stevenage, UK) or non-targeting ‘dummy’ antibody, delivered intratracheally in the same way as described for terbutaline.
Statistics
Model assumptions of normality were determined by analysis of residuals by Shapiro-Wilk test. Non-normally distributed data were transformed to normality where possible. Normally distributed data were evaluated by two-way analysis of variance (time course analysis) and Student’s t-test (endpoint studies). Non-normally distributed data were evaluated by Mann-Whitney U test or Kruskal-Wallis test followed by Dunn’s multiple comparisons test.
Results
LPS-induced lung injury model
Intranasal LPS induced weight loss peaking at 48 hours before returning towards pre-injury levels (figure 1A). At each time point, respiratory system elastance and arterial oxygenation were determined after 30 min of 8 mL/kg VT ventilation (with 100% O2) to standardise measurements. Elastance (figure 1B) increased during the course of injury. Arterial pO2 (figure 1C) was relatively normal at 24 hours after LPS but fell thereafter, attaining levels at 48–72 hours consistent with mild–moderate clinical ARDS (~200 mm Hg). Alveolar epithelial barrier permeability, evaluated by lavage fluid protein was increased throughout, peaking at 48–72 hours (figure 1D).

Time course of lipopolysaccharide-induced lung injury. Body weight (A) is expressed as % increase from pre-injured levels. Elastance (Ers) (B) and arterial oxygenation (PaO2/FiO2) (C) were evaluated following a 30 min period of non-injurious ventilation. Total protein (D) was determined in lung lavage fluid. N=4–6 for each readout at each time point, so all were treated as non-normally distributed data. Data are displayed as individual points with median and IQR, and were analysed by Kruskal-Wallis test followed by Dunn’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001 versus 0 hour (untreated mice). Exact p values were as follows: Panel A—24 hours, p=0.0134, 48 hours, p=0.0005, 72 hours, p=0.0890, 96 hours, p=0.3379; Panel B—24 hours, p=0.5720, 48 hours, p=0.0789, 72 hours, p=0.2730, 96 hours, p=0.0033; Panel C—24 hours, p>0.9999, 48 hours, p=0.0377, 72 hours, p=0.0146, 96 hours, p=0.1839; Panel D— 24 hours, p=0.4495, 48 hours, p=0.0092, 72 hours, p=0.0024, 96 hours, p=0.5134.
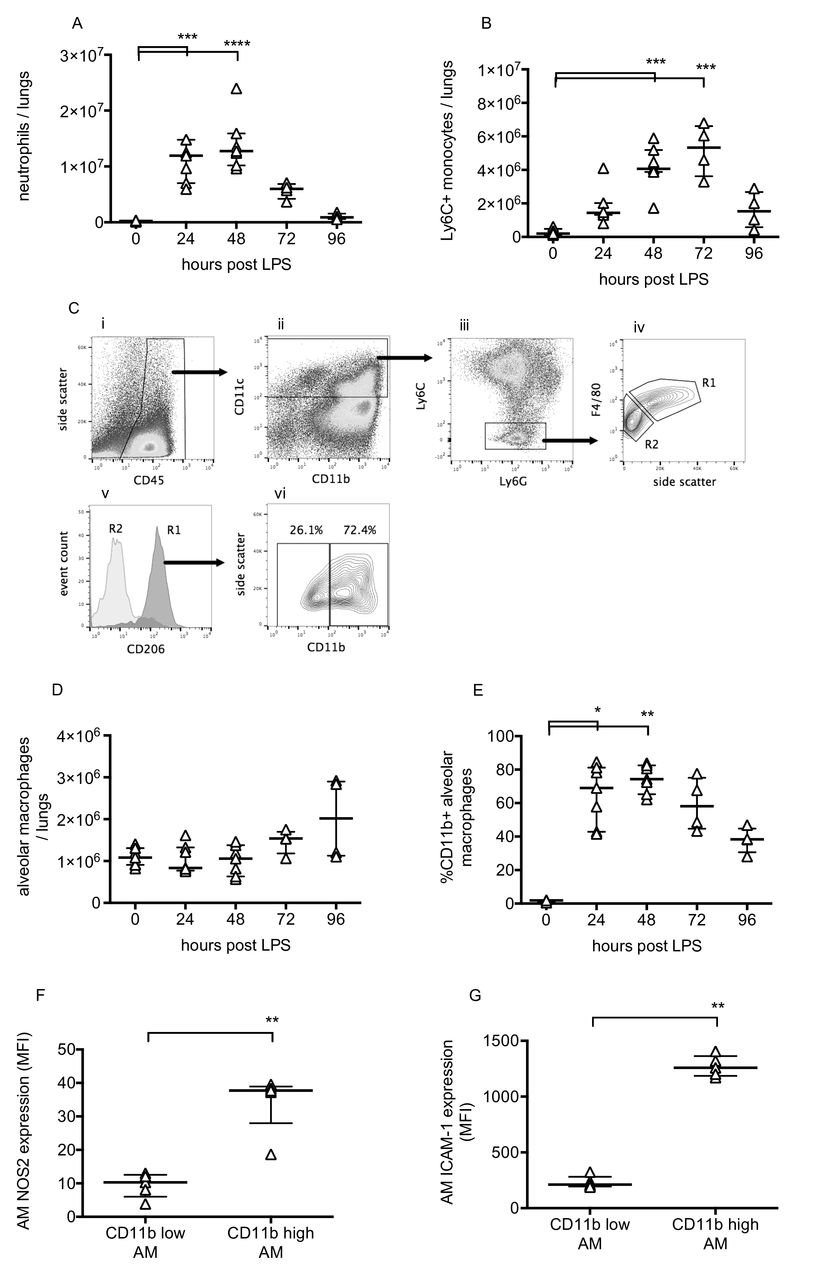
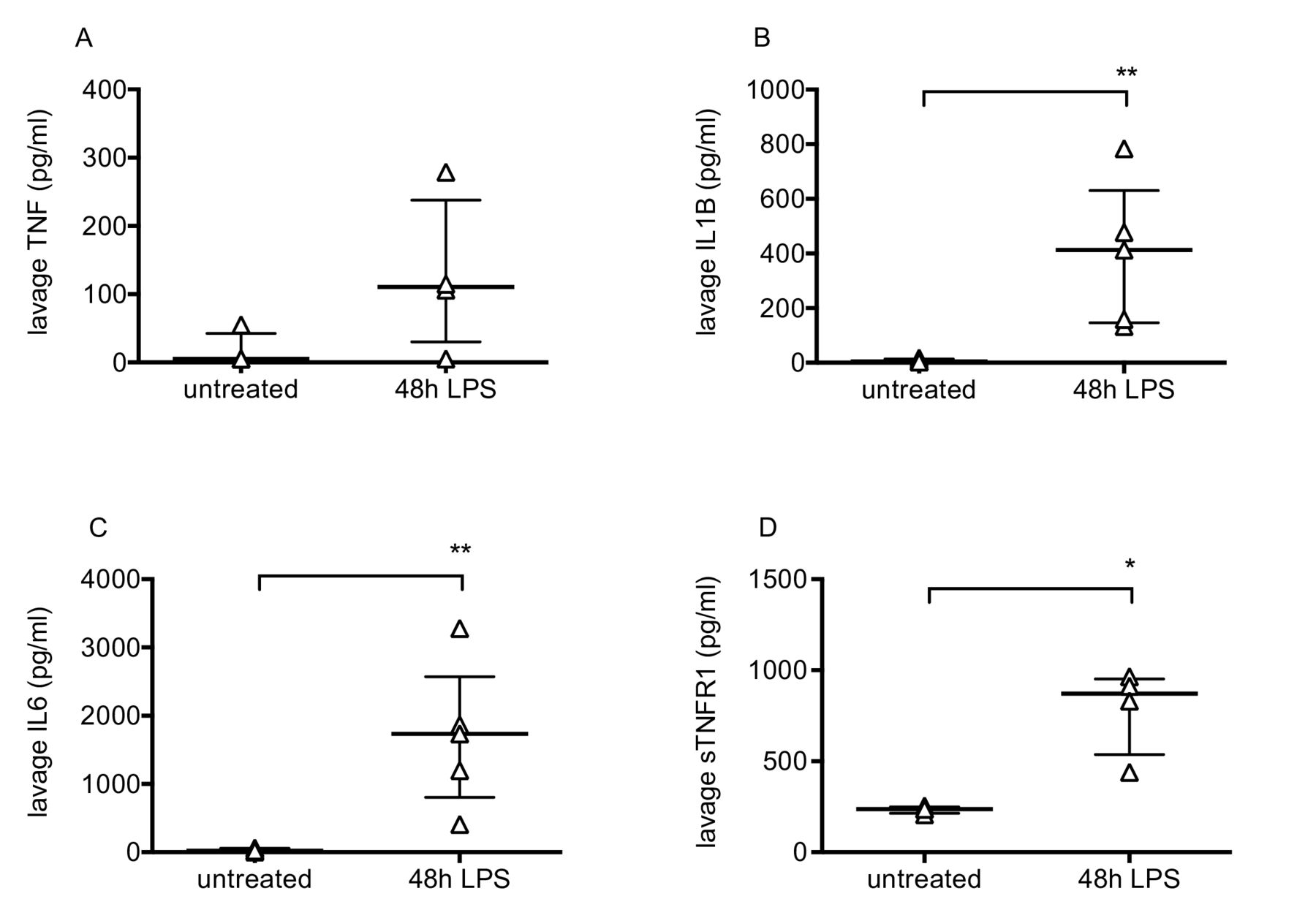
Pulmonary inflammation was determined by flow cytometric evaluation of leukocytes. Neutrophils and monocytes were defined as CD45+, CD11b+ and differentiated based on expression of Ly6C and Ly6G.11 Lung tissue neutrophil and Ly6C+ monocyte numbers changed dramatically during the progression of injury (figure 2A, B), peaking at 48–72 hours before returning to near normal levels. Alveolar macrophages were also investigated as they play a major role in the regulation of inflammation. These were defined as CD45+, CD11c+, Ly6C−, F4/80 high cells, and confirmed as having positive CD206 (mannose receptor) expression (figure 2C). While alveolar macrophage numbers did not change substantially (figure 2D), they dramatically upregulated their expression of CD11b, NOS2 and ICAM-1 (figure 2E–G). These data are consistent with previous findings12–14 suggesting polarisation of a subset of macrophages towards a more inflammatory phenotype. Finally, lavage fluid proinflammatory cytokines TNF, IL-1β and IL-6, along with soluble TNF receptor 1, were substantially elevated at 48 hours post-LPS (figure 3).
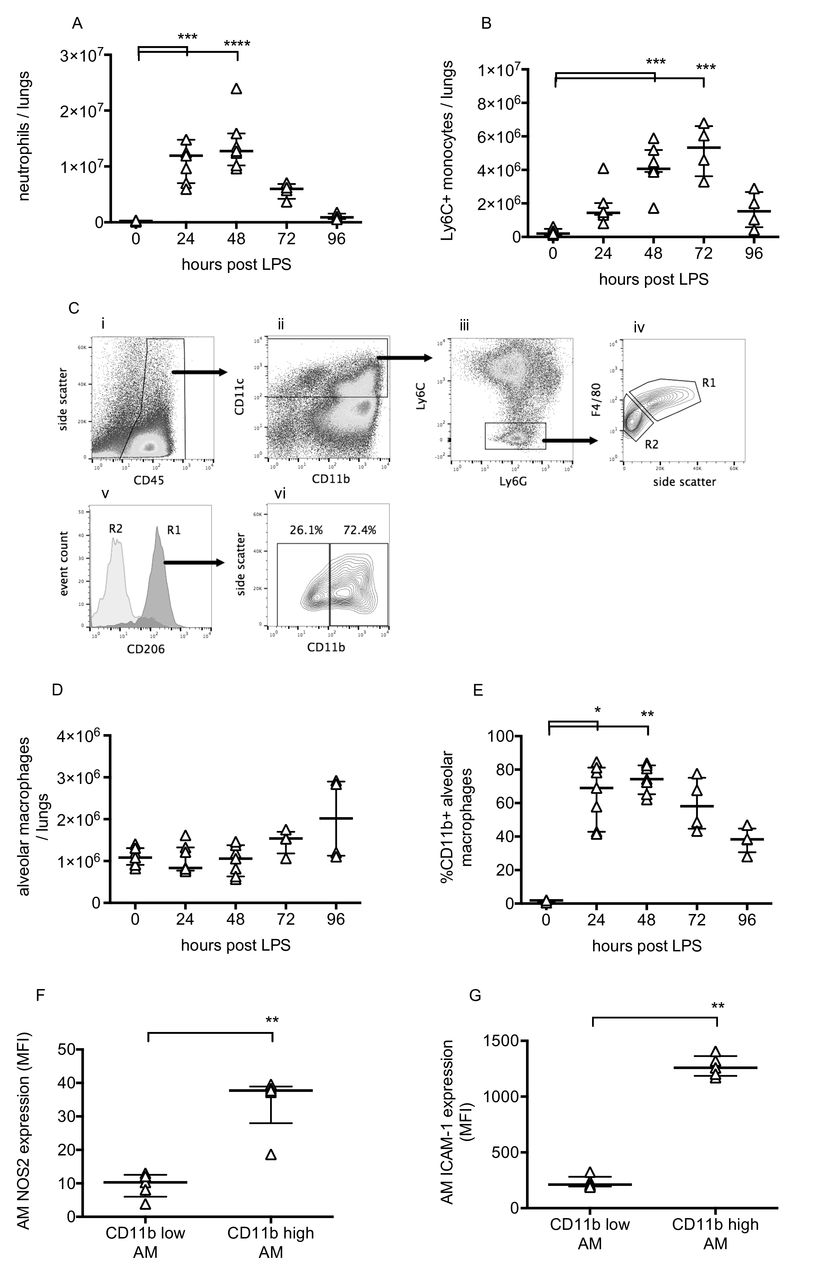
Leucocyte changes during LPS-induced lung injury. Lung tissue neutrophils (A) and Ly6C+ inflammatory monocytes (B) were quantified using flow cytometry of lung cell suspensions. Identification of resident alveolar macrophages in lungs 48 hours after LPS treatment is shown in Panel C. Macrophages were identified as CD45+ (Ci), CD11c+ (Cii), Ly6C and Ly6G low (Ciii) and F4/80 high (Civ, gate R1). Gate R1 cells were also confirmed as being CD206 (mannose receptor) positive (Cv), whereas no other cells were. Panel Cvi demonstrates that these cells, definitively identified as resident alveolar macrophages, existed in both CD11b-positive and CD11b-negative populations. Panel D shows alveolar macrophage number in lung tissue, and Panel E shows % of resident macrophages positive for CD11b expression. Expression of nitric oxide synthase 2 (Panel F) and ICAM-1 (Panel G) were evaluated in CD11b+ and CD11b− alveolar macrophages at 48 hours after LPS. N=4–7 for each readout at each time point, so all were treated as non-normally distributed data and are displayed as individual points with median and IQR. Data in Panels A, B, D and E were analysed by Kruskal-Wallis test followed by Dunn’s multiple comparisons test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 versus 0 hour (untreated mice). Data in Panels F and G were analysed by Mann-Whitney U test; **p<0.01. Exact p values were as follows: Panel A—24 hours, p=0.0008, 48 hours, p<0.0001, 72 hours, p=0.2191, 96 hours, p>0.9999; Panel B—24 hours, p=0.1213, 48 hours, p=0.0003, 72 hours, p=0.0003, 96 hours, p=0.4951; Panel D—24 hours, p>0.9999, 48 hours, p>0.9999, 72 hours, p=0.4571, 96 hours, p=0.4571; Panel E—24 hours, p=0.0121, 48 hours, p=0.0015, 72 hours, p=0.1059, 96 hours p>0.9999; Panel F, p=0.0079; Panel G, p=0.0079. LPS, lipopolysaccharide.

Intra-alveolar cytokines after LPS-induced lung injury. Bronchoalveolar lavage fluid levels of TNF (A), IL1-β (B), IL-6 (C) and soluble TNF receptor 1 (D) in untreated mice and mice 48 hours after intranasal LPS. N=4–5 for each group, so all were treated as non-normally distributed data. Data are displayed as individual points with median and IQR and were analysed by Mann-Whitney U test. *p<0.05, **p<0.01. Exact p values were as follows: Panel A, p=0.1429; Panel B, p=0.0079; Panel C, p=0.0079; Panel D, p=0.0286. IL, interleukin; LPS, lipopolysaccharide; TNF, tumour necrosis factor.
Effects of terbutaline in one-hit model of VILI
The impact of the β-agonist terbutaline was explored within the one-hit and two-hit models of VILI, specifically because β-agonists have shown contrasting outcomes in simple preclinical models versus patients with ARDS. The one-hit model was evaluated to confirm that the dose, route and timing of administration used would produce the anticipated protection within healthy lungs, based on previous studies.6 7 The initial respiratory system elastance was similar between the vehicle (saline) and terbutaline-treated groups (figure 4A) but as expected, terbutaline-treated animals were protected from the development of high VT (35 mL/kg) induced VILI. The overall increase in elastance (figure 4B) and deterioration in oxygenation (figure 4C) were substantially attenuated by terbutaline (p=0.003, p=0.006, respectively). As animals develop injury at different rates, we also evaluated the time course of changes in elastance and oxygenation (figure 4D, E), which were both attenuated by terbutaline (p<0.0001, p=0.0004, respectively). Finally, lavage fluid protein levels were also reduced (figure 4F; p=0.0041).

Physiological parameters during pure ventilator-induced lung injury model following terbutaline treatment. Initial elastance immediately after instillation and setting high VT was no different between saline and terbutaline-instilled animals (A). Panel B shows overall elastance change and Panel C shows final arterial oxygen levels in animals following ventilation with 35 mL/kg tidal volume (VT 35). As animals develop injury at different rates, time courses of elastance (D) and oxygenation (E) were also evaluated. Note that ‘end’ relates to the final value, either after 3 hours of ventilation or whenever animals met the mortality surrogate. Panel F shows protein concentration in lavage fluid taken at the end of experiments. N=6/group for all datasets. Endpoint data in Panels A, B and F were normally distributed, analysed by t-test and displayed as mean±SD. Data in Panel C required transformation before analysis and are displayed as back-transformed mean with 95% CIs. *p<0.05, **p<0.01 versus vehicle (saline) by Student’s t-test. Time course data in panel D were normally distributed and displayed as mean±SD while data in Panel E required transformation before analysis and are displayed as back-transformed mean with 95% CIs. Time courses were analysed by two-way analysis of variance. ***p<0.001, ****p<0.0001 for interaction. Exact p values were as follows: Panel A, p=0.1432; Panel B, p=0.0030; Panel C, p=0.0064; Panel D, p<0.0001 (interaction); Panel E, p=0.0004 (interaction); Panel F, p=0.0041.
Effects of terbutaline in two-hit model
To explore whether the two-hit model provided a closer approximation to the clinical scenario, we chose 48-hour post-LPS as our pre-injury, as this was where most parameters peaked in severity. While patients may not necessarily present at peak injury, we felt this best represented an ‘ARDS-like’ dysfunction that would necessitate ventilation. At this point, mice were instrumented and ventilated with a low (8 mL/kg) or moderate (20 mL/kg) VT. Importantly, neither LPS-injured animals ventilated with 8 mL/kg, nor healthy animals (without LPS injury) ventilated with 20 mL/kg VT displayed any worsening of respiratory mechanics or oxygenation (figure 5).

Time courses of elastance (A) and oxygenation (B) showed no deterioration in either 48 hours LPS-treated mice ventilated with 8 mL/kg (VT 8), or in healthy mice ventilated with 20 mL/kg (VT 20). N=6/group for both datasets. Data were normally distributed and displayed as mean±SD. LPS, lipopolysaccharide.
As with the one-hit model, the initial elastance following saline or terbutaline instillation was similar, indicating no immediate effect of the β-agonist within pre-injured lungs (figure 6A). As anticipated, LPS-injured mice instilled with saline and ventilated with 20 mL/kg displayed a substantial deterioration in elastance and oxygenation (figure 6B–E), demonstrating that pre-injury sensitised the lungs to moderate VT ventilation. Crucially, completely unlike the one-hit model, delivery of terbutaline into pre-injured lungs had no effect to attenuate secondary VILI, evaluated either by physiological parameters (figure 6B–E) or by lavage fluid protein (figure 6F). These data reflect the lack of effect of β-agonists in clinical ARDS,8 9 suggesting that this model was less likely to report a false-positive result than the simple one-hit model.

Physiological parameters following terbutaline treatment in two-hit lipopolysaccharide+ventilator-induced lung injury model. Initial elastance immediately after instillation and setting moderate VT (VT 20) was no different between saline and terbutaline-instilled animals (A). There was no impact of terbutaline treatment on respiratory mechanics or oxygenation, either in terms of overall endpoint changes (B and C) or time courses (D and E). Similarly, lavage fluid protein at the end of ventilation was no different (F). N=6/group for all datasets. Data were all normally distributed and displayed as mean±SD, except for Panel C which were non-normally distributed and displayed as box-whisker plot. Data were analysed by Student’s t-test (A, B and F), Mann-Whitney U test (C) and two-way analysis of variance (D and E) as appropriate. Exact p values were as follows: Panel A, p=0.3798; Panel B, p=0.4580; Panel C, p=0.2381; Panel D, p=0.3711 (interaction); Panel E, p=0.3102 (interaction); Panel F, p=0.7526.
Effects of TNF receptor 1 blockade in two-hit model of VILI
To evaluate whether the two-hit model was attenuable with a different pharmacological approach, mice with pre-existing lung injury were randomly instilled with either TNF receptor 1-targeting dAb or dummy non-targeting antibody, and ventilated using 20 mL/kg VT. We have previously shown that this antibody attenuates progression of injury when delivered prophylactically into healthy lungs using various one-hit models.15 16 Again, elastance immediately after drug instillation (figure 7A) was no different between groups (p=0.4858). Ventilation of dummy-treated mice with 20 mL/kg VT induced substantial deterioration of elastance and oxygenation, but unlike the situation with terbutaline, this secondary VILI was attenuated by TNF receptor 1 blockade. While there was not a major difference (figure 7B, C) in endpoint elastance (p=0.0712) or oxygenation (p=0.0906) following dAb treatment, time course evaluation (figure 7D, E) indicated clear attenuation of injury progression (p=0.0089 for elastance, p=0.0399 for oxygenation). Finally, TNF receptor 1 blockade reduced protein permeability (figure 7F, p=0.0295).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Physiological parameters following tumour necrosis factor receptor 1 dAb treatment in two-hit LPS+VILI model. Initial elastance immediately after instillation and setting moderate VT was no different between groups (A). Overall elastance change (B) and final oxygenation (C) showed trends towards improvement with dAb treatment (p<0.1) in mice ventilated with 20 mL/kg VT post-LPS. Time courses of elastance change (D) and oxygenation (E) were both significantly attenuated, as was final lavage fluid protein (F). N=6–7/group for all datasets. Data were all normally distributed and displayed as mean±SD, except for Panel C which were non-normally distributed and displayed as box-whisker plot. Endpoint data were analysed by Student’s t-test (A, B, F) or Mann-Whitney U test (C). *p<0.05 versus vehicle (dummy). Time course data (D and E) were analysed by two-way analysis of variance. *p<0.05, **p<0.01 for interaction. Exact p values were as follows: Panel A, p=0.4858; Panel B, p=0.0712; Panel C, p=0.0906; Panel D, p=0.0089 (interaction); Panel E, p=0.0399 (interaction); Panel F, p=0.0295. dAb, domain antibody; LPS, lipopolysaccharide; VILI, ventilator-induced lung injury.
Discussion
Despite identification of many therapeutic targets in preclinical models, clinically effective pharmacological treatments for ARDS have remained elusive. This does not mean that animal models are without value—indeed, the development of lung protective ventilation, the primary ‘therapy’ for patients with ARDS, was based ultimately on animal studies. However, it seems clear that while current models are useful for hypothesis generation and exploring biological processes, they appear suboptimal to predict outcomes of pharmacological intervention. Here, we describe a model which we feel contains the major components of a predictive scenario. We then explored a potential framework within which to determine the validity of this, and other models.
Due to the heterogeneous nature of ARDS aetiology, a range of preclinical models of varying complexity have been developed. While such variety of models is doubtless useful for exploring aspects of pathobiology, and while none could be thought perfect under all conditions, we would suggest that models intending to predict clinical efficacy should incorporate two particular considerations. First, models should incorporate mechanical ventilation. This is effectively obligatory for patients with ARDS, and thus any predictions which do not consider possible interactions between pharmacological intervention and ventilation are likely lacking. Second, models must use therapeutic dosing into injured lungs. The environment within which agents act in injured lungs is entirely different from that within healthy lungs. The differences are likely to be numerous, but we would envisage three crucial factors. First, alveolar flooding and congestion could be anticipated to impede delivery of inhaled drugs to the site of injury while intravenous delivery may be complicated by unpredictable regional blood flow distribution.17 Second, major changes in the environment within injured lungs, including increases in proinflammatory cytokines, leucocyte influx and changes in the phenotype of resident cells, could all influence the outcome of manipulating pathways. Finally, pre-existing injury leads to initiation of mechanisms which may desensitise pathways either in a protective way, for example, upregulation of endogenous antagonists and receptor shedding, or as a result of cell damage. Such a consideration was proposed to explain the ‘failure’ of a recent clinical trial of keratinocyte growth factor (KGF) in patients with ARDS18 despite it being beneficial in a range of animal and human preclinical models.19 Crucially, preclinical studies informing the trial demonstrated efficacy of KGF when used as a pretreatment or very early (1 hour) post-injury treatment.19–21 Ultimately, authors concluded that KGF may be ineffective in patients as receptors that are present in healthy lungs may not be present on injured epithelium.18 The presence or absence of pre-existing injury will certainly therefore influence the biology of pharmacological agents, and we should not expect similar outcomes. Moreover, modulation of pathways that play important roles in the initiating phases of ARDS may be counterproductive or even deleterious in established disease.
In the current study, we attempted to develop and validate a model which incorporates the important considerations outlined above. We chose to induce injury by intranasal administration of LPS, as a direct pulmonary injury/pneumonia-type model. Based on initial development studies, we utilised a time point of 48 hours post-LPS for our two-hit injury model, when mice most resembled a clinical picture of ARDS. They had lost weight, respiratory system elastance was increased, arterial oxygenation deteriorated and alveolar epithelial barrier permeability was increased. There were also dramatic changes in pulmonary leucocyte population and soluble pro-inflammatory/anti-inflammatory milieu. Onto this, we superimposed a relatively moderate (for a mouse) VT of 20 mL/kg, which in agreement with previous data,10 did not induce injury within healthy mice over 3 hours. Here however, when applied to mice with established injury, this protocol induced substantial worsening of lung function, indicating sensitisation of the lungs to ventilation. It is worth pointing out that the model does have limitations. Bolus administration of a very high dose of LPS was used as a reproducible, well-studied model but is unlikely to occur clinically. Additionally, the VT used was much higher than would be used in patients, partly as a reflection of the different respiratory mechanics between species.10 However, the model does allow us to explore the impact of pharmacological intervention within an environment that displayed heightened sensitivity to ventilation and was dramatically deviated from normal, both in terms of physiology and inflammation, as is the case within patients with ARDS.
We propose that such a ‘substantial deviation from normal’ should be a crucial aspect of models designed to predict or inform clinical use of pharmacological targets, and should be distinguished from more subtle scenarios. For example, a number of studies have utilised two-hit models of intrapulmonary LPS plus ventilation, but have initiated ventilation acutely (<4 hour) after LPS22–27 which may be informative but is unlikely to reproduce the biological derangement occurring within ARDS. Even with longer-term (up to 24 hours) LPS exposure, our data and that of others suggest that deterioration of important physiological parameters may not yet be present.28 29 In studies which explicitly claim that lungs were actually injured before ventilation was initiated,30–34 the primary marker evaluated is lavage fluid protein. However, this alone does not prove a physiologically meaningful lung injury.35 Thus, without clear evaluation of the degree of injury, most experimental paradigms exploring biological/immunological mechanisms of ARDS likely use regimes equivalent to very early intervention delivered into effectively healthy animals.3
While logic implies that better prediction of therapeutic outcomes would be achieved within the context of pre-existing injury, it remains difficult to prove this idea as there are no specific biomarkers for VILI or ARDS progression against which a model can be judged. We propose that the best current way to address this is to test a model against interventions which have been evaluated clinically. Here, we took the approach of exploring β-agonist therapy, specifically because this has produced disparate outcomes in clinical trials versus simple preclinical models. β-agonists improve alveolar fluid clearance, reduce permeability and attenuate oedema in models of acute Pseudomonas aeruginosa pneumonia,4 hyperoxia5 and VILI.6 7 However, both systemic and intrapulmonary β-agonists were ineffective in clinical trials.8 9 A model with better predictive power should therefore mirror the lack of efficacy seen clinically, rather than the false-positive findings of simple models. Here, we showed that while intratracheal terbutaline reduced injury in a one-hit pure VILI model as expected, it was ineffective when delivered into lungs with established injury. One suggestion for the failure of clinical β-agonist therapy is that the inflammatory environment within patients with ARDS may cause β-receptor desensitisation.36 Regardless of the mechanism underlying the differences in efficacy between healthy and pre-injured lungs, it is clear that the two-hit model produced a much less ‘misleading’ result compared with simpler models. It is worth clarifying though that for other scenarios, for example, ventilation of surgical patients without underlying injury, predictions from the pure VILI model may be more valid.
Finally, but importantly, we showed that the lack of efficacy of terbutaline within the two-hit model was not simply because the model was too injurious to be amenable to treatment. We have previously shown efficacy of prophylactic TNF receptor 1 inhibition using dAbs in models of VILI and acid aspiration,15 16 but here demonstrate that this remains effective when delivered into pre-injured lungs. Interestingly, this occurred despite the finding that soluble TNF receptor 1 levels were increased in lavage fluid following LPS (figure 3). It is not clear whether this soluble TNF receptor 1 upregulation was due to cell-surface receptor shedding37 or non-specific translocation from plasma38 but the efficacy of TNF receptor 1 inhibition indicates that receptors remained present on crucial target cells. The mechanisms by which TNF receptor 1 inhibition works remain unclear, but the fact that it was effective within a model which could not be attenuated by β-agonist therapy demonstrates that the mode of action of the two pharmacological agents are different, and that TNF signalling remains important during the progression of VILI even in the context of established injury.
In conclusion, a major reason for the lack of bench to bedside translation in ARDS research is likely to be misplaced confidence in targets. This comes partly from simple models using prophylactic administration of agents, as well as conflation of the concepts of statistical significance and biological importance. There has been much focus on development of models, but in our opinion a greater question relates to their validation. Developing more models without a system in which they can be judged is likely to perpetuate the current stagnation. We would propose a framework wherein knowledge of failed clinical pharmacologics represents an opportunity to validate new models and targets. A good model will be amenable to treatment but provide predictions of failure where that has been shown to happen clinically. A good target molecule will outperform a failed pharmacologic within a good model. We believe that the model described herein fulfils the criteria of a good model in that it incorporates crucial components of ventilation and pre-existing injury, and provided a better prediction of clinical β-agonist outcome than observed in simpler models. Given the heterogeneity of ARDS, we would not claim that this particular model would be the most appropriate under all conditions. Indeed, it may be considered one step on a pathway including varying strains, sex, age and comorbidities of animals to better reflect the human situation.3 While this will have substantial resource implications, moving quickly from simple models to clinical trials seems to be a false economy. We believe that adopting a framework, wherein new models and targets are evaluated alongside interventions with a known clinical outcome, would represent a crucial step towards reducing the likelihood of ineffective treatments being tested in patients.
Acknowledgments
The authors would like to acknowledge the support and input from Biopharm Molecular Discovery, GlaxoSmithKline R&D, in particular Dr Joanna Cordy, Dr Tracey Wright, Dr Peter Morley and Dr Andrew Bayliffe.
References
Footnotes
Contributors Conception and design; CMO, KPO, MT, MRW. Data collection, analysis and interpretation; CMO, MWK, SS, RFB, MRW. All authors participated in manuscript revisions and approved the final version.
Funding This study was supported by funding from GlaxoSmithKline (STU100031322; STU3000031807) and the Biotechnology and Biological Sciences Research Council (BB/L502261; BB/R505274/1). SS was recipient of a clinical research training fellowship from the Medical Research Council/British Journal of Anaesthesia (MR/M018164/1). MWK is supported by the Agency for Science, Technology and Research (A*STAR) NSS (MBBS-PhD) scholarship.
Competing interests MW and MT received research funding from GlaxoSmithKline to carry out this work. GSK have a financial interest in the use of domain antibodies, including those targeting p55 TNF receptor, in the treatment of pulmonary and other diseases. GSK were involved in the initial design of the study but had no other involvement in collection, analysis and interpretation of data, or in the decision to submit the report for publication.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.