Article Text

Abstract
Rationale Goblet cell hyperplasia (GCH) is one of the cardinal features of chronic obstructive pulmonary disease (COPD) and contributes to airways obstruction. Rhinovirus (RV), which causes acute exacerbations in patients with COPD, also causes prolonged airways obstruction. Previously, we showed that RV enhances mucin gene expression and increases goblet cell number in a COPD mouse model. This study examines whether RV causes sustained GCH in relevant models of COPD.
Methods Mucociliary-differentiated COPD and normal airway epithelial cell cultures and mice with normal or COPD phenotype were infected with RV or sham and examined for GCH by immunofluorescence and/or mucin gene expression. In some experiments, RV-infected COPD cells and mice with COPD phenotype were treated with γ-secretase inhibitor or interleukin-13 neutralising antibody and assessed for GCH. To determine the contribution of NOTCH1/3 in RV-induced GCH, COPD cells transduced with NOTCH1/3 shRNA were used.
Results RV-infected COPD, but not normal cell cultures, showed sustained GCH and increased mucin genes expression. Microarray analysis indicated increased expression of NOTCH1, NOTCH3 and HEY1 only in RV-infected COPD cells. Blocking NOTCH3, but not NOTCH1, attenuated RV-induced GCH in vitro. Inhibition of NOTCH signalling by γ-secretase inhibitor, but not neutralising antibody to IL-13, abrogated RV-induced GCH and mucin gene expression.
Conclusions RV induces sustained GCH via NOTCH3 particularly in COPD cells or mice with COPD phenotype. This may be one of the mechanisms that may contribute to RV-induced prolonged airways obstruction in COPD.
- airway epithelium
- viral infection
- copd pathology
Statistics from Altmetric.com
Key messages
What is the key question?
Does rhinovirus infection induce goblet cell hyperplasia in chronic obstructive pulmonary disease (COPD) and what is the underlying mechanism?
What is the bottom line?
Rhinovirus, which is associated with acute exacerbations in COPD, can promote goblet cell hyperplasia, one of the features of airway epithelial remodelling in COPD.
Why read on?
We demonstrated that rhinovirus induces goblet cell hyperplasia in in vitro and in vivo models of COPD through NOTCH signalling independent of well-characterised interleukin-13 pathway.
Introduction
Mucus overlaying the airway epithelium traps the inhaled pathogens and other noxious agents and the coordinated beating of cilia clear mucus along with the trapped inhaled agents. Thus, both cilia and mucus contribute to primary defence mechanism of airway epithelium. However, in chronic inflammatory airway disease including chronic obstructive pulmonary disease (COPD), there is excessive mucus production, which may subsequently cause airways obstruction and disease exacerbation. Excessive production is a result of increased synthesis and secretion of mucins and is often associated with increase in number of goblet cells (goblet cell hyperplasia or GCH). Patients with COPD show GCH in both small and large airways, and this may be a result of abnormal responses to infections or other noxious agents.
Rhinovirus (RV), which causes self-limiting upper airway infections in healthy subjects, is associated with acute exacerbations in COPD. Patients with mild COPD experimentally infected with RV show more severe and prolonged lower airway symptoms including airflow obstruction.1 We demonstrated that RV enhances mucin gene expression and GCH within 4 days postinfection in two different models of COPD.2 3 Therefore, it is plausible that RV may induce or perpetuate GCH in patients with COPD, thus causing excessive mucus production and airways obstruction. However, mechanisms by which RV induces sustained GCH in COPD are not well understood.
Most studies conducted on respiratory virus-induced GCH have focused on models of allergic asthma or virus infection model that induces type II responses. Although viral infection causes airway obstruction in both patients with asthma and COPD, the underlying mechanisms may differ because of different type of responses. In allergic asthma, viral infection predominantly induces type 2 cytokines, such as interleukin (IL)-13, IL-4 and IL-5.4 IL-13 has been shown to play a critical role in inducing GCH via activation of STAT6, SPDEF and FOXA3.5–7 Therefore, most of the published studies on allergic asthma have focused on delineating the molecular mechanisms by which respiratory viruses induce IL-13.8–11 In COPD, however, viral infection is associated predominantly with CXCL-8, CXCL-10, TNF-α, IFN-γ and IL-6.12 Therefore, the role of IL-13 in virus-induced GCH in COPD is not clear.
Another mechanism by which respiratory viruses may induce GCH is via activation of epidermal growth factor receptor (EGFR). Activation of EGFR has been shown to induce GCH independently or in concert with IL-13. For instance, persistent activation of EGFR in mucociliary-differentiated cell cultures by treatment with reactive oxygen species has been demonstrated to reduce number of ciliated cells and increase goblet cells.13 Chronic EGFR activation by Sendai virus induces GCH in well-differentiated airway epithelial cells by promoting survival and transdifferentiation of ciliated to goblet cells in concert with IL-13.14 Recently, we demonstrated that RV promotes goblet cell differentiation in a model of injured but not in normal airway epithelium via persistent activation of EGFR.15 Since EGFR activity is already enhanced in COPD,16 17 RV may further activate EGFR to drive GCH independently or in concert with IL-13.
In recent years, NOTCH signalling has emerged as one of the key players in regulating goblet cell differentiation in conductive airways. NOTCH signalling pathway is evolutionarily conserved, and therefore all mammals express NOTCH receptors and ligands. There are four NOTCH receptors (NOTCH1–4) and five NOTCH ligands (delta-like ligand (DLL) 1, 2, 3, 4, and jagged (JAG) 1 and 2). Human bronchial epithelial cells express all four NOTCH receptors and all NOTCH ligands except for DLL3.18 Overexpression of active domain of either NOTCH 1 or 3, but not 2 or 4, skewed differentiation of cells towards secretory cells with a parallel decrease in ciliated cells. Similarly, overexpression of NOTCH ligand JAG 1 also increased goblet cell differentiation of human airway basal cells19 and in mice.20 In murine system, the NOTCH signalling increases during repair of injured airway epithelium. However, persistent activation of NOTCH signalling was associated with increase in goblet cell number with concomitant decrease in ciliated cells.21 Consistent with these findings, inhibition of NOTCH signalling with antagonist, γ-secretase inhibitor promoted ciliated cell differentiation and decreased number of goblet cells in explants of mouse trachea22 and in primary human tracheobronchial epithelial cell cultures.23 In contrast to these findings, in developing lungs, inactivation of pofut1, a transcription factor activated by NOTCH signalling, led to overabundance of goblet cells in trachea.24 In an allergic mouse model of asthma, and in respiratory syncytial virus-infected mice, neutralisation of NOTCH ligand, DLL4, produced by dendritic cells induced GCH via enhancement of IL-13 expression by T cells.25 26 Since both activation and inhibition of NOTCH signalling appear to play a role in development of GCH, it is plausible that cellular response to NOTCH signalling may depend on cell type, whether the cells are proliferative or postmitotic, type of infection and underlying lung disease. However, the contribution of NOTCH signalling in RV-induced GCH in COPD is yet to be determined.
In the present study, we investigated whether RV induces sustained GCH and also determined the contribution of IL-13, EGFR and NOTCH signalling pathway in RV-induced GCH in relevant in vitro and in vivo models of COPD.
Materials and methods
Cell cultures
Collection of tracheobronchial segments at the time of double lung transplantation from both COPD recipients and donor lungs for isolation of airway basal cells was approved by the Institutional Review Board, University of Michigan. Age and clinical status of COPD subjects and healthy donors are provided in online supplementary table 1. Airway basal cells at passage one were cultured at air/liquid interface as described previously to promote mucociliary differentiation.15 27 For details, please see the supplemental file. The sample size for COPD and normal cell cultures was determined based on our previous study,28 in which we examined the goblet cell number. With the calculated effect size of 2.87, α=0.05 and power of 0.95, we determined that we will require cells from six normal subjects and six subjects with COPD. Therefore, we used cells from 8 to 9 patients in our initial experiments to determine the difference in responses to RV between normal and COPD cell cultures. The sample size required for detecting significant differences between sham and RV infection in COPD cultures was determined based on the results of initial experiments (figure 2D), in which we quantified GCH induced by RV. With the effect size of 3.89, α=0.05 and power of 0.95, we determined that we will require cells from three normal subjects and three subjects with COPD. Therefore, we used cells from 3– to 6 subjects to determine the mechanism by which RV induces GCH in COPD cells.
Supplementary file 1
Rhinovirus
RV16 and RV1B purchased from ATCC were propagated in H1 HeLa cells, partially purified, and viral titre was determined by plaque assay as described.29 Less than 100 kDa fraction from purified RV preparation was used as sham control. All in vitro experiments were conducted with major group RV RV16 and in vivo studies with minor group virus RV1B, because major group viruses do not infect mouse airways. Moreover, previously, RV1B has been shown to stimulate similar innate immune responses as RV16 in human airway epithelial cells.29
Infection of cell cultures
Mucociliary-differentiated cell cultures were infected apically with 10 µL of phosphate buffered saline (PBS) containing RV16 at 1 multiplicity of infection (MOI) or equivalent volume of sham and incubated for 15 days as described previously.15 Medium in the basolateral chamber was changed every other day. In some experiments, cells were treated basolaterally with erlotinib, or γ-secretase inhibitor N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) every other day starting from 2 days post-RV infection. Some infected cultures were treated either basolaterally or apically with neutralising antibody to IL-13 or normal IgG every other day for 15 days. In some experiments, cells were pretreated with erlotinib daily for 7 days and infected with sham or RV, and cultures were maintained as above. Dimethyl sulfoxide (DMSO)- or normal IgG-treated cells were used as negative controls as appropriate. Cytotoxicity of these agents was determined by assessing lactate dehydrogenase levels in the basolateral medium.
PAS-Alcian blue staining
Five micron-thick sections of paraffin embedded cell cultures were deparaffinised and stained with PAS/Alcian blue to detect goblet cells.
Transduction of primary human airway epithelial cells
Primary human airway epithelial cells were transduced with GIPZ lentiviral human NOTCH1 or NOTCH3 shRNA or control shRNA during the first week of culturing as described previously.15
Quantitation of goblet cells
Number of goblet cells in cell cultures was determined as described previously.15 Briefly, after relevant treatment, apical surface of the cell cultures was washed with 0.15% sodium bicarbonate in PBS, fixed in ice cold methanol and then immunostained with antibody to human tracheobronchial mucins30 and imaged under confocal microscopy to detect goblet cells. The nuclei were detected with DAPI. Number of goblet cells per 0.1 mm2 were counted in 25 random fields per cell culture and averaged.
RV infection of mice
Animal use and protocols were approved by Institutional Animal Care and Use Committee at the University of Michigan. Female C57BL/6 mice (6–8 weeks old) were used in these experiments. Mice were housed in specific pathogen-free facility. Normal mice or mice with mild COPD phenotype were briefly anaesthetised with isoflurane and infected with 50 µL of 1×108 plaque forming unit (PFU)/mL RV1B by intranasal route.2 Mice were then treated every other day with 50 µL of gamma secretase inhibitor (GSI) L685,458, a γ-secretase inhibitor by intranasal route. We used GSI L685,458 in mice, because DAPT that was used in the cell culture system was not well tolerated by mice. Mice were sacrificed at 8 days postinfection; lungs were harvested for isolation of total RNA or fixed in paraformaldehyde and embedded in paraffin. Paraffin sections were stained with periodic Schiff’s reagent to detect goblet cells. In agreement with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines, we determined the required number of animals per group by power analysis using the data from our previous study2 in which we examined the effect of RV on expression of Muc5AC in mice with COPD phenotype. With the calculated effect size of 2.45, α=0.05 and power of 0.95, we determined that we will require a minimum of five animals per group. Therefore, we used six animals per group in most of the experiments to achieve significance.
Western blot analysis
After relevant treatment, total protein was extracted, and equal amount of protein was subjected to Western blot analysis with antibodies to NOTCH1, NOTCH3, HEY1 or β-actin. Specific bands were semiquantified by densitometry using NIH ImageJ and expressed as fold change over β-actin.
Gene arrays and real-time PCR
Normal and COPD cells infected with sham or RV were harvested at 15 days postinfection. Total RNA was isolated, and purified RNA was subjected to gene array analysis using affymetrix Human Gene ST 2.1. Microarrays and bioinformatics analysis was performed by microarray core facility and bioinformatics core facility, respectively, at University of Michigan. Difference in the expression of genes between sham and RV were defined as >2 fold change with p value <0.05.
After relevant treatment, total RNA was isolated from airway epithelial cells or mouse lungs, and mRNA expression of NOTCH1, NOTCH3, MUC5AC, MUC5B, FOXA3 and other genes was determined by using gene-specific primers and normalised to housekeeping gene, β-actin (for mice) or glyceraldehyde 3-phosphate dehydrogenase (for cells).
Statistical data analyses
All cell culture experiments were conducted three times in duplicates or triplicates. Average from three independent experiments for each subject was calculated and used in further statistical analysis. Animal experiments were conducted two times with three mice per group for a total of six mice per group. To pinpoint statistical significance, non-parametric analyses, such as the exact Wilcoxon rank-sum test for comparing two independent groups and the signed-rank test to compare paired samples between the groups and intra-comparison between sham and RV-infected cultures within a group, were used. A p value of <0.05 was considered statistically significant, and the exact p values are provided in the figures. SAS V.9.4 was used for all the data analyses. Per cent of inhibition was calculated considering 100% for DMSO-treated or NTshRNA-transfected samples and expressed as % inhibition±SD.
Results
RV induces persistent increase in mucin gene expression in COPD airway epithelial cell cultures
COPD and normal airway epithelial cell cultures infected with RV or sham were maintained for 15 days, and the expression of mucin genes, MUC5B and MUC5AC, was determined. Sham-infected COPD cell cultures showed higher expression of both MUC5B and MUC5AC than similarly infected normal cultures as observed previously28 (figure 1A,B). Mucin gene expression in COPD cell cultures negatively correlated with percent predicted FEV1 of patients indicating that excessive mucus production may indicate loss of lung function (online supplementary figure 1). Expression of both MUC5AC and MUC5B genes further increased in RV-infected COPD cell cultures but not in normal cell cultures (figure 1C,D).

RV infection enhances mucin gene expression in COPD, but not in normal airway epithelial cell cultures. Mucociliary-differentiated normal and COPD airway epithelial cell cultures were established from airway basal cells obtained from nine healthy non-smokers and eight subjects with COPD in duplicates. Two cultures from each subject was infected with sham and another two with RV. At 15 days postinfection, total RNA was isolated, and mucin gene expression was determined by qPCR. Expression of mucin genes was normalised to housekeeping gene, G3PDH. Average from three independent experiments for each subject was calculated and presented as range with median. (A and B) Comparison of sham-infected normal and COPD cultures (the exact Wilcoxon rank sum test). (C and D) Within-group paired comparison of mucin gene expression between sham-infected and RV-infected normal and COPD cultures (the signed-rank test). COPD, chronic obstructive pulmonary disease; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; RV, rhinovirus.
Next, we examined whether enhanced mucin gene expression parallels increase in goblet cells. PAS/Alcian blue staining of paraffin-embedded cell culture sections and immunostaining of whole cultures with tracheobronchial mucins suggested presence of more goblet cells in COPD than in normal cell cultures as observed previously28 (figure 2A,B). RV-infected COPD, but not normal cell cultures, showed further increase in goblet cells. Number of goblet cells per 0.1 mm2 was higher in COPD than in normal cultures (figure 2C). After RV infection, COPD, but not normal cells, showed further increase in number of goblet cells compared with respective sham-infected cell cultures (figure 2D, intracomparison). This was associated with increased expression of FOXA3, a transcription factor expressed in goblet cells at basal levels (sham infected) and as well as after RV infection in COPD cells (figure 2E,F). Basal expression levels of FOXA2, a transcription factor that negatively regulates goblet cell differentiation, and FOXJ1, which promotes ciliated cell differentiation, were relatively lower in COPD than in normal cell cultures, while there was no change in the expression of SPDEF, SCGB1A1, a marker of club cells (online supplementary figure 2A, C, E and G). RV infection had no significant effect on the expression of FOXJ1, FOXA2, SPDEF or SCGB1A1 (online supplementary figure 2B, D, F and H). These results indicate that RV-induced mucin gene expression parallels with GCH, which further enhances in COPD cells with no effect on either ciliated cells or club cells.

RV increases number of goblet cells in COPD cell cultures. Mucociliary-differentiated airway epithelial cell cultures were established from nine normal subjects and eight subjects with COPD. Two cultures from each subject were infected with sham and another two with RV and maintained for 15 days. (A) Paraffin sections of cell culture were stained with PAS/Alcian blue to detect goblet cells by histology (arrows represent goblet cells). (B) Apical surface of cell cultures was washed to remove secreted mucin, fixed in methanol, and goblet cells were detected by using antibody to tracheobronchial mucins followed by confocal microscopy (red: goblet cells, and blue: nuclei). (C and D) Number of goblet cells per 0.1 mm2 were counted in 25 random fields from each culture, and the counts from duplicate cultures were averaged. (E and F) Total RNA was isolated and mRNA expression of FOXA3 was determined by qPCR. Figure parts C and E show difference in number of goblet cells and FOXA3 expression, respectively, between normal and COPD cultures at basal level (the exact Wilcoxon rank-sum test). (D and F) Intracomparison of number of goblet cells and FOXA3 expression, respectively, between sham-infected and RV-infected normal and COPD cultures (the signed-rank test). Data represent average from three independent experiments calculated for each subject. COPD, chronic obstructive pulmonary disease; RV, rhinovirus.
RV-induced GCH is independent of IL-13
To determine whether RV-induces GCH through IL-13, we determined the expression of IL-13 mRNA by qPCR and protein by ELISA. There was no detectable levels of IL-13 protein in either normal or COPD airway epithelial cells irrespective of infection. Both normal and COPD cells expressed very low levels of IL-13 mRNA, which did not change after RV infection (online supplementary figure 3A). Additionally, treatment with neutralising antibody to IL-13 from basolateral side did not reduce either the number of goblet cells or the expression of mucin genes (online supplementary figure 3B–D). Similar results were observed when cell cultures were treated with antibody to IL-13 from the apical side. To confirm the efficacy of IL-13 neutralisation antibody, normal cell cultures were cultured in the presence of IL-13 to induce GCH and then treated with neutralising antibody to IL-13 or isotype control and examined for expression of MUC5AC and goblet cell number. IL-13 induced GCH and expression of MUC5AC and SPDEF (online supplementary figure 3E–H). Neutralising antibody to IL-13 inhibited MUC5AC and SPDEF mRNA and goblet cell number in IL-13 treated cultures (online supplementary figure 3I–K) confirming the efficacy of IL-13-neutralising antibody. These results indicate that IL-13 may not contribute to RV-induced GCH in COPD airway epithelial cells under the current experimental conditions.
Blocking of EGFR activity does not inhibit RV-induced GCH in COPD
Next, we examined the role of EGFR in RV-induced mucin gene expression or GCH in COPD cells. Treatment of COPD airway epithelial cell cultures with 1 µM erlotinib for 14 days reduced activity of EGFR as assessed by EGFR phosphorylation on tyrosine residue 1173 without causing cytotoxicity as observed previously17 (online supplementary figure 4A,B). Furthermore, sham-infected and RV-infected COPD cultures showed similar EGFR phosphorylation. Cell cultures treated with 2.5 µM and 5 µM erlotinib showed cytotoxicity, and very few cells were left after 14 days of treatment irrespective of infection, and therefore EGFR activity was not determined. The observed cytotoxicity at 2.5 µM and 5 µM may be due incremental accumulation of the drug in the cells as a result of repeated treatment. Based on these results, subsequent experiments were conducted with 1 µM erlotinib. COPD airway epithelial cell cultures were infected with sham or RV, and then treated with DMSO or 1 µM erlotinib on alternate days starting from 2 days postinfection. Erlotinib appears to reduce expression of both MUC5B and MUC5AC in sham-infected cells without altering expression of FOXA3 or goblet cell number (figure 3A–D). However, erlotinib neither attenuated mucin gene expression nor goblet cell number or FOXA3 expression in RV-infected cell cultures. Additionally, histological evaluation of RV-infected cells did not indicate reduction in goblet cells (figure 3E). To examine whether inhibition of EGFR prior to infection prevents RV-induced GCH, cell cultures were pretreated with erlotinib for 7 days and infected with sham or RV and mucin gene expression, and GCH was assessed. Again, erlotinib had no effect on RV-induced expression of mucin genes or GCH (online supplementary figure 5A–C). These results indicate that although EGFR may play a role in promoting mucin gene expression in uninfected COPD cells, it does not contribute to RV-stimulated GCH or mucin gene expression in these cells.

EGFR signalling does not contribute to RV-induced GCH in COPD. Mucociliary-differentiated airway epithelial cell cultures were established from six subjects with COPD in quadriplicates. Four cultures from each subject were treated with sham and another four with RV. Two sham and two RV-infected culture from each subject was treated with 1 µM of erlotinib, and another identical set was treated with DMSO (vehicle control) for 14 days. Experiment was repeated three times with duplicates or triplicates, and data were averaged for each subject. (A to C) Total RNA was isolated, and the expression levels of mucin genes and FOXA3 were determined by qPCR. (D) From an identical experiment, cells were fixed and immunostained with antibody to tracheobronchial mucins to quantify goblet cells as described under figure 2. Data represent intracomparison between sham and RV and comparison between RV-infected DMSO and erlotinib treated wells (the signed-rank test, paired comparison). (E) Paraffin sections of the cell cultures were stained with PAS/Alcian blue to detect goblet cells by histology (arrows represent goblet cells) and images are representative of six COPD cultures. COPD, chronic obstructive pulmonary disease; RV, rhinovirus.
Microarray analysis indicates a role for Notch signalling pathway in RV-induced GCH in COPD cell cultures
In an attempt to identify the potential mechanism underlying RV-induced GCH and mucin gene expression, we subjected RV-infected or sham-infected COPD and normal airway epithelial cell cultures to microarray array analysis. Out of 48 226 genes, 97 and 107 genes were differentially (two fold or higher) (online supplementary tables 2 and 3) expressed with p value of <0.05 in RV-infected normal and COPD cells, respectively, compared with respective sham controls. In normal cells, out of 97 differentially expressed genes, 30 genes were upregulated and 64 genes were downregulated. In RV-infected COPD cells, out of 107 differentially expressed genes, 78 genes were upregulated and 26 genes were downregulated. Ingenuity pathway analysis indicated activation of NOTCH signalling pathway in RV-infected COPD cells with upregulation in the expression of NOTCH1, NOTCH3 and downstream effector gene HEY1. RV-infected normal cells showed downregulation of NOTCH1 and NOTCH3 with no change in HEY1. Assessment of mRNA expression by qPCR indicated increased expression of NOTCH1, NOTCH3 and HEY1 in RV-infected COPD but not in normal cells (figure 4A–C). At protein level, the expression of only NOTCH3 and HEY1 was increased in RV-infected COPD cells when compared with sham-infected cells (figure 4D–G). Interestingly, RV-infected normal cells showed significant decrease in the expression NOTCH1 and NOTCH3 at mRNA level with no change at protein level. Surprisingly, normal cells had no detectable levels of HEY1 protein irrespective of infection; therefore, it was expressed as ‘0’ in figure 4G. Furthermore, qPCR assessment indicated increased mRNA expression of NOTCH ligands JAG1 and JAG2, but not DLL1, DLL2 or DLL4 (online supplementary figure 6) in RV-infected COPD cells. Normal cells, however, showed significant decrease in all the measured NOTCH ligands except for DLL4, NOTCH2 and HEYL following RV infection. There was no difference in the expression of NOTCH4, and effector proteins, HES1 and HES5 after RV infection in either normal or COPD cells (online supplementary figure 6F–J).

RV upregulates expression levels of NOTCH1, NOTCH3 and HEY1 in COPD cell cultures. (A to C) Mucociliary-differentiated cells cultures established from nine normal subjects and eight subjects with COPD were infected with sham or RV, and after 15 days, expression levels of NOTCH1, NOTCH3 and HEY1 were determined by qPCR. Data from three independent experiments were averaged and presented as intracomparison between sham and RV (paired analysis, the signed-rank test). (D) From an identical experiment, total protein was isolated and subjected to Western blot analysis with antibodies to NOTCH1, NOTCH3, HEY1 and β-actin. Images are representative of cells obtained from nine normal subjects and eight subjects with COPD. Line in bottom right panel of figure part G indicates that image was compiled from two different blots developed on the same day under identical conditions. N1, N3, N8 and C6, C7 and C9 are cells obtained from three normal subjects and three subjects with COPD, respectively. COPD, chronic obstructive pulmonary disease; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; RV, rhinovirus.
To examine the contribution of NOTCH signalling in RV-induced mucin gene expression and GCH, COPD cell cultures were infected with RV or sham and then treated with γ-secretase inhibitor, DAPT, which has been shown to inhibit NOTCH signalling.18 Initial experiments with varying concentrations of DAPT indicated that DAPT at 10 µM effectively blocks RV-induced HEY1 expression without causing cytotoxicity in COPD airway epithelial cell cultures (online supplementary figure 7). Therefore, we used DAPT at 10 µM in subsequent experiments. Treatment with DAPT reduced RV-induced GCH in COPD epithelial cell cultures as assessed by PAS/Alcian blue staining (figure 5A). DAPT significantly reduced number of goblet cells and the expression of FOXA3, MUC5AC, MUC5B and HEY1 in RV-infected cultures (figure 5B–F). Compared with RV-infected DMSO treated cultures, similarly infected DAPT treated cultures showed substantial reduction in goblet cells and mRNA levels of FOXA3, MUC5AC, MUC5B and HEY1 (table 1). DAPT had no effect on the expression of Club cell marker SCGB1A1 (online supplementary figure 7C).

DAPT blocks RV-induced changes in COPD cell cultures. Mucociliary-differentiated COPD cultures established from cells obtained from eight subjects with COPD were infected with sham or RV and maintained for 15 days. Cells were treated with DMSO (vehicle) or DAPT on every other day starting from 2 days postinfection. (A) Paraffin sections of RV-infected cultures treated with DMSO or DAPT were stained with PAS/Alcian blue, representative images from eight subjects with COPD from each group (arrows represent goblet cells). (B) From an identical experiment, cells were fixed and immunostained with antibody of tracheobronchial mucins to quantify goblet cells. (C–F) Total RNA was extracted and expression levels of FOXA3, MUC5AC, MUC5B and HEY1 were determined by qPCR. Data are averaged from three independent experiments and presented as within-group paired comparison between sham-infected and RV-infected DMSO and DAPT-treated cultures and comparison between RV-infected DMSO and DAPT-treated groups (the signed-rank test). COPD, chronic obstructive pulmonary disease; G3DPH, glyceraldehyde 3-phosphate dehydrogenase; RV, rhinovirus.
DAPT inhibits RV-induced goblet cell metaplasia in COPD cell cultures
COPD cells transduced with non-targeting (NT) shRNA or shRNA specific to NOTCH1 or NOTCH3 lentivectors were used to confirm the contribution of specific NOTCH receptor in RV-induced GCH. Compared with NT shRNA-transduced cells, NOTCH3, but not cells transduced with NOTCH1, showed reduced goblet cells following RV infection (figure 6A,B and table 2) and attenuated expression of FOXA3, MUC5B, MUC5AC and HEY1 (figure 6C–F and table 2). Western blot analysis confirmed the inhibition NOTCH1 and NOTCH3 expression by respective shRNA (figure 6G,H). These results indicate that RV may induce GCH in COPD airway epithelial cell cultures by stimulating NOTCH3 signalling.

Genetic inhibition of NOTCH3, but not NOTCH1, abrogates RV-induced GCH in COPD cell cultures. Cells obtained from four patients with COPD were transduced with either non-targeting (NT) shRNA, NOTCH1 shRNA or NOTCH3 shRNA and cultured at air/liquid interface to promote mucociliary differentiation. Two cultures from each shRNA group were infected with sham and another two with RV and incubated for 15 days. (A) RV-infected cultures were assessed by histology to detect goblet cells, representative images from four COPD cultures each from RV-infected groups (arrows indicate goblet cells). (B) Goblet cells were quantified by immunofluorescence confocal microscopy. (C–F) Total RNA was isolated, and the expression levels of FOXA3, MUC5B, MUC5AC and HEY1 were determined by qPCR, normalised to G3PDH. Data represent within-group paired comparison between sham-infected and RV-infected cultures derived from three independent experiments carried out in duplicates or triplicates (the signed-rank test). Paired comparison of RV-infected cultures between NT shRNA and NOTCH1 shRNA or NOTCH3 shRNA groups was also conducted (the signed-rank test). (G and H) Representative Western blots showing inhibition of NOTCH1 and NOTCH3 expression by respective shRNA. COPD, chronic obstructive pulmonary disease; G3PDH, glyceraldehyde 3-phosphate dehydrogenase; RV, rhinovirus.
Inhibition of NOTCH3 abrogates of RV-induced goblet cell metaplasia in COPD cell cultures
RV-infected mice with COPD phenotype show increased expression of Notch3 and Hey1
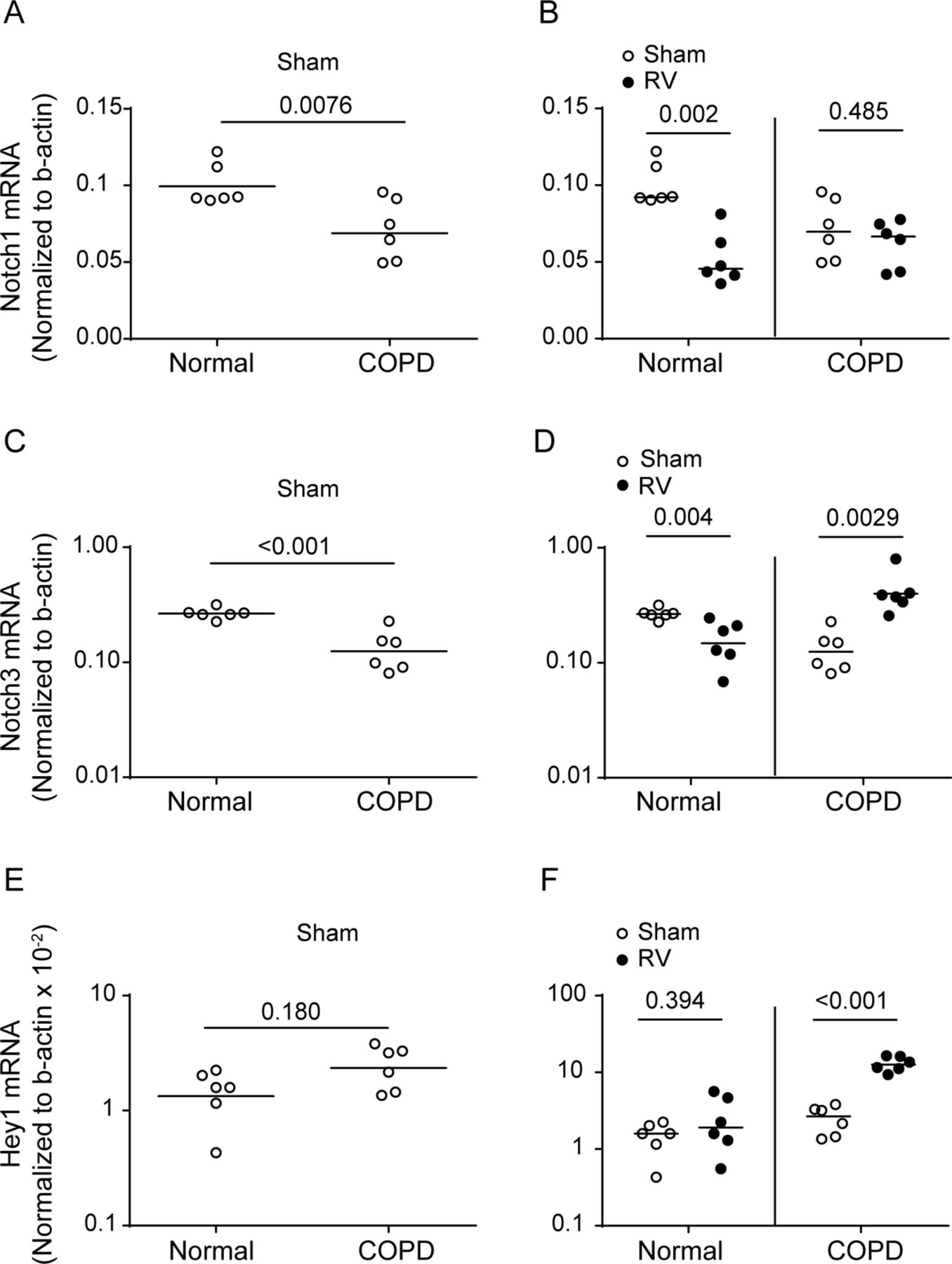
Previously, we demonstrated that RV increases GCH and mucin gene expression in mice with COPD phenotype at 4 days postinfection.2 In the present study, we found that mice with COPD phenotype infected with RV show sustained GCH up to 8 days following viral infection (figure 7A). As observed previously, mice with COPD phenotype showed increased expression of goblet cell markers, Foxa3 and Muc5AC 2 (figure 7B,D). Expression of these genes further increased following RV infection (figure 7C,E). Interestingly, normal mice showed higher expression of Notch1 and Notch3 than mice with COPD phenotype at basal levels (figure 8). Following RV infection, normal mice showed significant decrease in both Notch1 and Notch3 mRNA expression and no difference in Hey1 expression. In contrast, mice with COPD phenotype showed increase in the expression of Notch3 and Hey-1, but not Notch1. Additionally, compared with RV-infected normal mice, similarly infected mice with COPD-phenotype showed significantly higher mRNA levels of Notch3 and Hey1 but not Notch1.
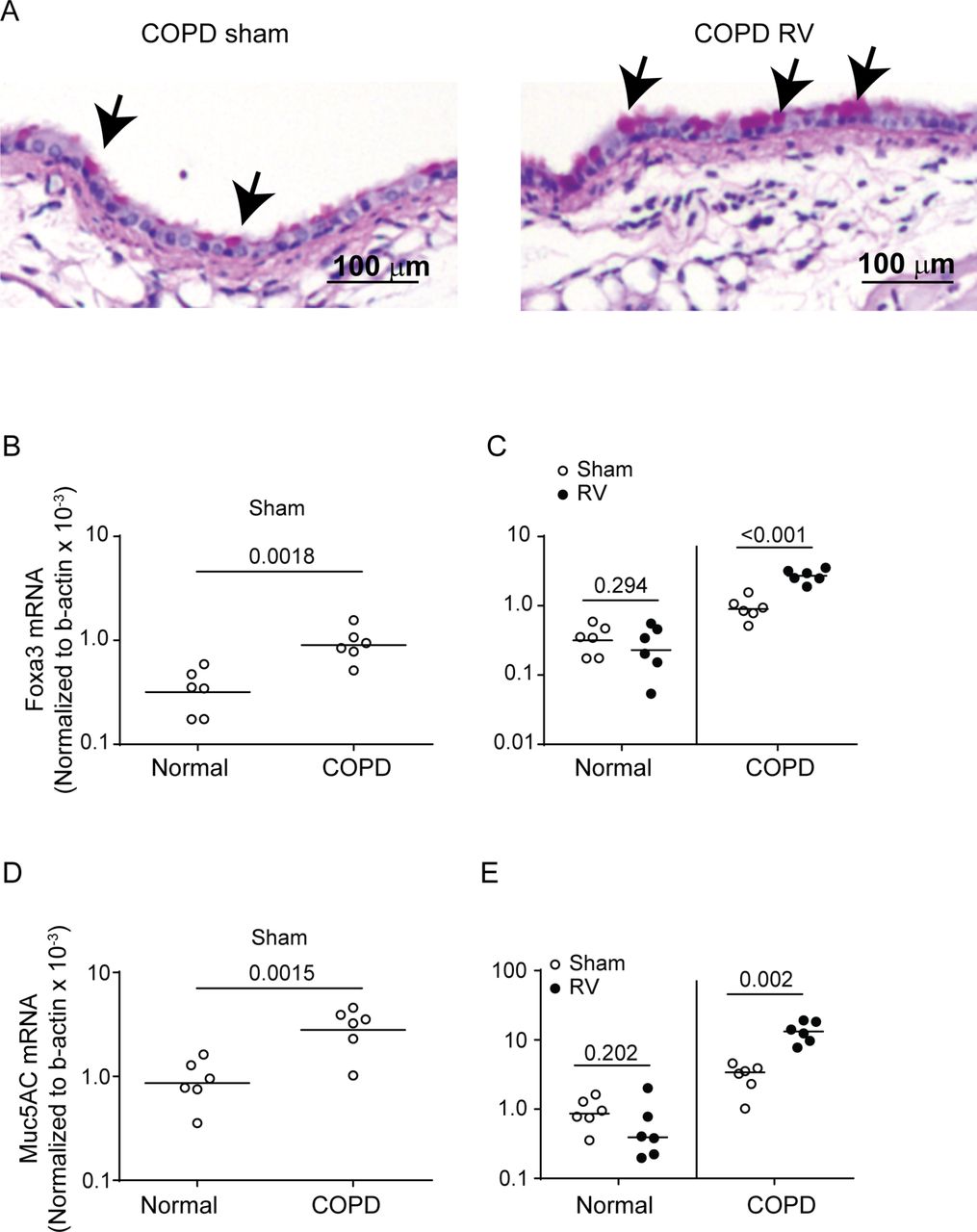
RV-infected mice with COPD phenotype show sustained GCH and expression of Foxa3 and Muc5AC. Normal mice or mice with COPD phenotype were infected with sham or RV1B and sacrificed after 8 days. (A) Lungs were fixed, embedded in paraffin, and lung sections were stained with PAS to detect goblet cells (arrows represents goblet cells). (B–E) Total RNA was isolated, lung mRNA levels of Foxa3 and Muc5AC were determined by qPCR and data were normalised to β-actin. Data represent range with median. (B and D) Comparison between sham-infected normal mice and sham-infected mice with COPD phenotype (the exact Wilcoxon rank sum test). C and E, comparison between sham and RV infection for normal mice and mice with COPD phenotype (the exact Wilcoxon rank-sum test). Experiment was conducted two times with three mice per group (total six mice per group). COPD, chronic obstructive pulmonary disease; GCH, goblet cell hyperplasia; PAS, Periodic acid-Schiff’s stain; RV, rhinovirus.

RV stimulates expression of Notch3 and Hey1 in mice with COPD phenotype. Normal mice or mice with COPD phenotype were infected with either sham or RV1B and sacrificed after 8 days. Total RNA was isolated, and lung mRNA levels of Notch1, Notch3 and Hey1 were determined by qPCR. Data were normalised to β-actin and presented as range with median. (A, C and E) Comparison between sham-infected normal mice and sham-infected mice with COPD phenotype, and (B, D and F) comparison between sham and RV infection within each group (the exact Wilcoxon rank-sum test). Experiment was conducted two times with three mice per group (total six mice per group). COPD, chronic obstructive pulmonary disease; RV, rhinovirus.
To examine the contribution of Notch signalling in RV-induced GCH in vivo, mice with COPD phenotype were treated with γ-secretase inhibitor, GSI L685,458 (0.003, 0.03 and 0.3 mg/kg body weight) on alternate days up to 8 days postinfection (four treatments) and assessed for expression of FoxA3, Muc5AC and Hey1 by qPCR (figure 9). GSI L685,458 at 0.03 and 0.3 mg/kg body weight significantly inhibited RV-induced Foxa3 and Muc5AC, and this was associated with reduced expression of Hey1 indicating NOTCH signalling plays a role in RV-induced GCH in vivo. Dose-dependent effect of GSI L685,458 on RV-induced expression of FoxA3, Muc5AC and Hey1 is shown in table 3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of Notch abrogates RV-induced GCH in mice with COPD phenotype. Mice with COPD phenotype were infected with sham or RV1B and then treated with γ-secretase inhibitor GSI L685,458 (0.003, 0.03 or 0.3 mg/kg body weight) or DMSO (vehicle control) by intranasal route every other day for 8 days. (A–C) Lung mRNA levels of Hey1, Foxa3 and Muc5AC were determined by qPCR. Data represent range with median (n=5–6 mice per group; the exact Wilcoxon rank-sum test). COPD, chronic obstructive pulmonary disease; GCH, goblet cell hyperplasia; RV, rhinovirus.
GSI L685,458 inhibits RV-induced expression of FoxA3, Muc5AC and Hey1 in a dose-dependent manner
Although airway epithelial cells do not express IL-13, other cell types in the lung may produce IL-13, which can then stimulate IL-13 receptor-dependent GCH development in the airways. Therefore, we determined the role of IL-13 in RV-induced GCH in vivo. First, we assessed the expression of IL-13 by qPCR. Compared with normal, mice with COPD phenotype showed increasing trend in the expression of IL-13 in sham-infected mice (online supplementary figure 8A). After RV infection, however, mice with COPD phenotype showed decreasing trend in IL-13 compared with sham-infected mice, which did not reach significance. Furthermore, treatment with IL-13 neutralising antibody following RV infection had no effect on RV-induced Foxa3 or Muc5AC (online supplementary figure 8B,C) indicating IL-13 may not contribute to RV-induced GCH in vivo.
Taken together, our results indicate that RV induces sustained mucin gene expression particularly in COPD airway epithelial cell cultures, and this was accompanied by increase in number of goblet cells by approximately 1.7 fold. In contrast, RV had no significant effect on the expression of markers of club and ciliated cells. NOTCH3, which is upregulated by RV, was found to contribute significantly to RV-induced GCH and mucin gene expression.
Discussion
This study highlights one of the mechanisms by which RV may induce mucus production and possibly airways obstruction in COPD. As far as we know, this is the first report demonstrating that RV, which does not cause long-term pathological changes in normal airway epithelium, enhances GCH and mucin gene expression in COPD airway epithelial cell cultures and in mice with COPD phenotype. Interestingly, we found that RV-induces GCH and mucin gene expression through NOTCH activation, but not via activation of IL-13 or EGFR in relevant models of COPD.
RV is associated with acute exacerbations in COPD. Additionally, RV has been shown to persist longer and cause sustained lower respiratory symptoms including airways obstruction after experimental infection in patients with COPD.1 These findings suggest that RV, which was originally thought to only cause acute infection, may actually cause long-lasting pathological changes including remodelling of airway epithelium. This study provides first in vitro and in vivo experimental evidence that RV infection enhances/induces GCH, one of the important features of remodelled airway epithelium, in relevant models of COPD. Although small amounts of viral RNA was recovered up to 7 days, no live virus was detected from the lungs of mice with COPD phenotype beyond 4 days after infection (Comstock and Sajjan, unpublished results) implying that altered and/or persistent host responses may be responsible for the observed GCH.
Airway epithelial cells respond to exogenously added IL-13 by showing increased mucin gene expression and development of GCH in vitro via activation of NOTCH2.31 Recently, upregulation of IL-13 expression following IL-33 release from airway epithelium was shown to increase GCH in mice infected with Sendai virus.32 Since elevation in IL-33 was associated with IL-13 and goblet cells in severe COPD, the authors speculated a role for IL-13 in inducing GCH following viral infection in COPD. In the present study, however, RV did not induce expression of IL-13 or NOTCH2 either in COPD cells or in mice with COPD phenotype (mice exposed to cigarette smoke). Additionally, neutralisation of IL-13 had no effect on RV-induced GCH in both in vivo and in vitro models of COPD. These results are consistent with previously published observations in which influenza virus did not induce IL-13 expression in cigarette smoke-exposed mice despite increased expression of IL-33.33 These observations suggest that IL-13, which contributes to development of GCH in experimental models of asthma and Sendai virus infection, may not play a role in RV-induced GCH in relevant models of COPD.
EGFR expression is already elevated in COPD bronchial epithelium. Since RV activates EGFR and induces acute expression of MUC5AC in normal epithelial cells,34 35 and GCH in repairing airway epithelial cell cultures,15 we reasoned that RV-induced EGFR may contribute to the observed GCH in COPD. To our surprise, RV did not increase EGFR phosphorylation in COPD cells. Furthermore, EGFR inhibition neither attenuated mucin gene expression nor reduced GCH induced by RV. These observations indicate that EGFR may not contribute to RV-induced GCH in COPD.22
NOTCH signalling, which plays a pivotal role in cell differentiation during lung development, also regulates cell differentiation in adult airways.18–23 31 Intriguingly, gene array analysis identified increased mRNA expression of NOTCH1, NOTCH3 and the effector gene HEY1 in RV-infected COPD cells indicating activation of NOTCH signalling in RV-infected COPD cells. Inhibition of NOTCH signalling significantly attenuated RV-induced GCH and the expression of FOXA3 and mucin genes in COPD cells and in mice with COPD phenotype. Previous studies suggest that both NOTCH1 and NOTCH3 promote cell differentiation of airway basal stem cells to secretory cell lineage.18 36 Furthermore, while NOTCH3 was shown to be required for transitioning of airway basal cells to differentiating progenitors, NOTCH1 and NOTCH2 were necessary for promoting differentiation of progenitors into secretory cells during regeneration of normal airway epithelium in vitro.36 In the current study, although expression of both NOTCH1 and NOTCH3 was significantly elevated at mRNA levels, only NOTCH3 protein expression was elevated in RV-infected COPD cells. Furthermore, mice with COPD phenotype also showed increase in Notch3 but not Notch1 expression after RV infection. Additionally, genetic inhibition of NOTCH3, but not NOTCH1, abrogated RV-induced GCH and expression of FOXA3 and mucin genes in COPD airway epithelial cell cultures. These observations clearly indicate a role of NOTCH3 in RV-induced GCH in COPD cells, but the precise mechanisms is yet to be investigated. Even though, activation of NOTCH3 is expected to decrease total protein levels of NOTCH3 as it gets processed by γ-secretase to low molecular weight active NICD3, we observed increased expression of NOTCH3 protein. This is not surprising because NOTCH, being a transcription factor, may stimulate its own expression. Consistent with this notion, increased expression of NOTCH1 protein was observed following NOTCH1 activation by Mycobacterium bovis in macrophages.37 These observations suggest that NOTCH3, but not NOTCH1, may contribute to RV-induced GCH in COPD.
Previous studies have reported that compared with normal, airway epithelial cells from smokers with or without COPD show reduced expression of NOTCH ligands and receptors.38 Consistent with this observation, we also observed comparatively lower expression of NOTCH3 in COPD airway epithelial cell cultures and in mice with COPD phenotype. Since NOTCH signalling is essential for differentiation of airway basal cells, such reduction in NOTCH may cause expansion of basal cells causing basal cell metaplasia that is often observed in patients with COPD.39 In line with this, our ongoing studies indicate presence of more airway basal cells in mucociliary-differentiated COPD cell cultures than in normal cell cultures (Comstock and Sajjan, unpublished data). Since RV infects airway basal cells readily,15 40 it is plausible that NOTCH3 expression induced by RV may promote differentiation of these basal cells to goblet cell lineage, thus driving GCH particularly in COPD. Since RV did not reduce markers of either ciliated or club cells, it is unlikely that RV-induced GCH occurs as a result of transdifferentiation of these cells. Further studies are under progress to determine whether COPD basal cells are more amenable to RV infection than normal cells, or availability of abundant basal cells renders COPD cultures more vulnerable to RV infection. In addition, we are also conducting studies to identify the relevant NOTCH ligand that is responsible for activation of NOTCH3 in RV-infected COPD cells.
Interestingly, mRNA expression of NOTCH1, NOTCH2, NOTCH3 and all the NOTCH ligands was significantly reduced in normal cells after RV infection. This may be one of the innate immune mechanisms that is required for maintenance of airway epithelial homeostasis after RV infection, which is lacking in COPD, thus, leading to enhanced GCH.
Although our in vivo studies indicate that RV-induced GCH and Hey1, a downstream effector of NOTCH, is effectively blocked by GSI L685,485, we cannot exclude the possibility of off target effects of this compound. GSI L685,485 blocks the function of infiltrated eosinophils in a mouse model of asthma.41 Therefore, it is possible that GSL685,485, in addition to blocking NOTCH signalling, may also inhibit function of other infiltrated cells in the lungs in response to RV, which may influence GCH development. Since genetic inhibition of NOTCH3 blocks RV-induced GCH in vitro, and GSI L685,485 abrogates RV-induced Hey1, we speculate that the observed effect of GSI L685,485 may be due NOTCH inhibition.
As observed previously, we found that cells from patients with COPD retain their ability to maintain in vivo phenotype in showing GCH and increased expression of mucin genes.28 Although EGFR inhibition attenuated mucin gene expression, it did not reduce GCH or FOXA3 expression in these cells. Furthermore, there was no difference in the expression of SPDEF between normal and COPD cultures. These observations suggest a role for signalling pathway leading to activation of FOXA3 that is independent of EGFR or SPDEF in the development of GCH in COPD airway epithelial cell cultures. Interestingly, mucin gene expression at basal levels negatively correlate with FEV1 of the donor, indicating the association of mucus production with loss of lung function. Although cells derived from COPD subjects, which maintain in vivo phenotype, are useful in delineating mechanisms of RV-induced pathological changes, further experiments with bronchial biopsies and/or sputum samples collected from COPD patients with acute exacerbations associated with RV are necessary to confirm these findings and will be pursued in the near future.
In summary, we provide evidence that RV, which only causes self-limiting upper airway infection in normal subjects, may induce GCH in COPD, one of the features of airway epithelial remodelling. Unlike in allergic airway disease where GCH occurs under the influence of IL-13, NOTCH signalling may play a role in promoting GCH, particularly after RV infection in COPD. However, further studies in COPD subjects with RV-associated exacerbations are needed to confirm our findings.
Acknowledgments
We would like to thank Dr Fernando Martinez, Cornell University, Ithaca, New York and Dr Catherine Meldrum, University of Michigan, Ann Arbor, Michigan, for helping us to obtain tracheobronchial segments from healthy and COPD subjects. We would also like to thank Microarray core facility at University of Michigan for performing microarrays on normal and COPD samples and assistance with data analysis.
References
Footnotes
Contributors YJ and JAG designed and performed experiments. RM, DP and ATM provided technical assistance with cell culture and animal work. DY performed statistical analysis and wrote description of statistical analysis. US conceived the idea for the manuscript, helped to design appropriate experiments and prepared the manuscript.
Funding This work was supported by National Institutes of Health grants HL897720, AT004793 and AT007620 to US.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Institutional regulatory board, University of Michigan, Ann Arbor, Michigan.
Provenance and peer review Not commissioned; externally peer reviewed.