Article Text

Statistics from Altmetric.com
Cyril Schweitzer (CS): A 1.5-year-old girl was referred to the Paediatric Pneumology Department of the University Hospital of Nancy, France, for progressive tachypnoea and hypoxia. She suffered from intrauterine growth retardation, and was born prematurely at 31 weeks’ gestation; birth weight: 1.160 kg (≤2 SD); birth length: 37 cm (≤2 SD). The patient presented with acute respiratory distress syndrome at birth, requiring treatment with a dose of surfactant, 9 days of mechanical ventilation and 8 weeks of oxygen therapy. On arrival to our centre, she was dyspnoeic on exertion and oxygen saturations were between 91% and 94%. Further investigations included a high-resolution CT scan and a bronchoalveolar lavage (BAL).
Laureline Berteloot (LB) and Julie Bruneau (JB): CT imaging of the chest revealed a ‘crazy paving’ pattern whereas analysis of BAL fluid failed to identify any underlying disease.
CS: Hypoxia transiently improved after initiation of inhaled corticosteroids and antireflux therapy but relapsed after a few months, so that a second BAL was performed.
JB: The second BAL fluid revealed the presence of extracellular lipid and protein deposits and foamy alveolar macrophages.
CS and Cécile Bonnet (CeB): These abnormalities led to a diagnosis of pulmonary alveolar proteinosis (PAP) at the age of 2.6 years, which was confirmed by genetic testing; complex chromosome abnormalities were detected, with (1) deletion of CSF2RA and CRLF2 on the X chromosome inherited from the mother, and (2) a de novo rearrangement of the paternal X chromosome, resulting in loss of both CSF2RA alleles.1
CS, Christophe Delacourt (CD) and Jacques De Blic (JDB): Recessive mutations in the α or β subunits of the granulocyte macrophage colony-stimulating factor receptor (GM-CSF-R, encoded respectively by CSF2RA and CSF2RB) cause two subtypes of hereditary PAP.2 Impairment of GM-CSF-dependent surfactant clearance by alveolar macrophages leads to the progressive accumulation of surfactant in the alveolar space resulting in hypoxemic respiratory failure.2 PAP is associated with high morbidity and mortality rates. Regular removal of excess surfactant via whole-lung lavage (WLL) is the standard of care for hereditary PAP.
CS: Despite the implementation of six monthly WLL, progression of the tachypnoea, exercise intolerance and hypoxia required intensification of the treatment regimen, with WLL every 3 weeks and continuous oxygen therapy from the age of 5 years onwards.
CS, CD, JDB: Given that PAP related to GM-CSF-R deficiency is haematopoietic by nature, haematopoietic stem cell transplantation (HSCT) from a healthy donor might be a curative option, as shown in a murine model.3 This case was discussed with colleagues from the Paediatric Immunology and Haematology Unit.
Despina Moshous (DM), Alain Fischer (AF), Stéphane Blanche (SB) and Bénédicte Neven (BN): Given the child’s severe condition, the poor prognosis and the putative curative potential of HSCT (ie, colonisation of the lungs with alveolar macrophages derived from a healthy donor), a transplantation with a 10/10 human leucocyte antigen (HLA)-matched unrelated male donor was proposed. The patient’s parents agreed to the procedure and provided written, informed consent.
Coralie Briand (CoB), Guilhem Cros (GC) and Marie-Louise Frémond (MLF): The patient was transferred to the Paediatric Immunology and Haematology Unit of Necker Hospital (Paris, France) at the age of 6.3 years. On arrival, her weight was 16 kg (−2 SD) and her height was 105 cm (−2 SD). She was tachypnoeic at rest (respiratory rate of 30/min), and, on continuous oxygen therapy of 1.5 L/min, oxygen saturations were 95%. Routine laboratory exams were normal and there was no evidence of active infection.
LB and Alice Hadchouel (AH): Last CT scan of the chest before HSCT is shown in figure 1. Lung function tests showed a slight, restrictive, ventilatory defect, with a functional residual capacity of 80% predicted and a moderate reduction in the diffusion capacity of the lung for carbon monoxide to 65% of the normal value.

{kind=link}
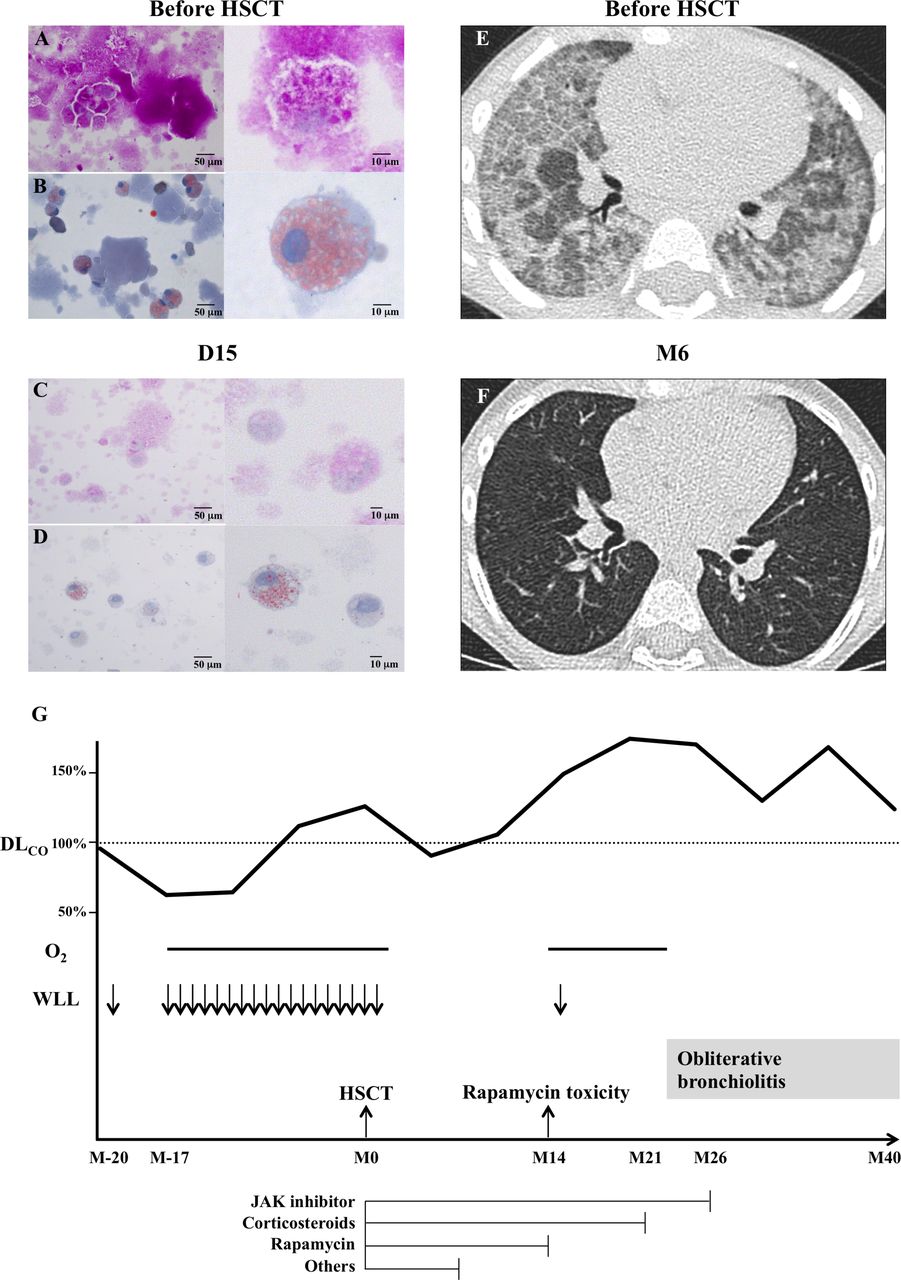
Histopathological analysis of the bronchoalveolar lavage (BAL) fluid before and after HSCT and reversal of pulmonary alveolar proteinosis (PAP) (according to a CT assessment) 6 months after HSCT, and treatment outline (A–D), BAL before HSCT (A and B) and at D15 afterwards (C and D), showing a reduction in the extracellular lipid and protein deposits and foamy alveolar macrophages (left panels: bar=50 µm; right panels: enlarged details, bar=10 µm; A and C: periodic acid-Schiff (PAS) staining; B and D: oil red O staining). (E) High-resolution CT scan of the chest before HSCT, showing a ‘crazy paving’ pattern consistent with PAP. (F) High-resolution CT scan of the chest 6 months (M6) after HSCT, demonstrating the absence of lesions. (G) The treatment timeline, from 20 months (M20) before HSCT to 40 months (M40) afterwards. DLCO, diffusion capacity of the lung for carbon monoxide; HSCT, haematopoietic stem cell transplantation; Janus kinase (JAK) inhibitor, ruxolitinib; O2, oxygen therapy; WLL, whole-lung lavage. ‘Others’ refer to cyclosporine, mycophenolate mofetil, antithymocyte globulin, inolimomab, rapamycin and infliximab.
DM, AF, SB and BN: Due to the poor condition of the child and the risk of the procedure, we recommended a reduced-intensity conditioning regimen consisting of intravenous busulfan 12.8 mg/kg with targeted cumulative area under the curve of 14 000–15 000 µM·min, fludarabine 160 mg/m2 and antithymocyte globulin 10 mg/kg. Graft-versus-host disease (GVHD) prophylaxis should consist of cyclosporine from day (D)−1 and mycophenolate mofetil from D0.
Alessandra Magnani (AM): Bone marrow from a 10/10 HLA-matched unrelated male donor was harvested. The graft contained 7.6×106 CD34+cells/kg.
CoB, GC and MLF: Aplasia was uneventful, and haematological recovery was achieved on D15 post-HSCT with 100% donor chimerism (according to an X and Y chromosome fluorescent in situ hybridisation analysis of peripheral blood). The patient’s respiratory status remained stable during aplasia and improved slightly as the time of haematological recovery.
JB: BAL on D15 revealed a drastic reduction in the numbers of extracellular lipid and protein deposits and foamy cells (figure 1C,D). Chimerism performed on BAL cells was 94% of donor origin.
CoB, GC and MLF: From D23, respiratory condition rapidly normalised allowing complete and long-lasting cessation of oxygen therapy. Skin (grade II) and gut (grade IV) acute GVHD occurred on D66. The latter required six successive lines of immunosuppressive drugs (figure 1G).
LB and AH: Despite life-threatening GVHD, long-term respiratory status improvement was documented in the patient. At 6 months after HSCT, she had a normal respiratory examination while lung CT scan and pulmonary function tests showed complete normalisation (figure 1F).
Cécile Pochon (CP): Fourteen months after HSCT, while on rapamycin, corticosteroids and ruxolitinib, tachypnoea, hypoxia and haemoptysis occurred so that the patient required continuous oxygen therapy. The patient was then transferred to the transplantation unit at Necker Hospital (Paris, France) for further investigations.
CoB, GC and MLF: Because of the long-lasting immunosuppression, we suspected a respiratory infection or a graft-related toxicity. We therefore performed a BAL and a lung biopsy.
JB: Analysis of the BAL fluid showed alveolar haemorrhage and, surprisingly, the lung biopsy displayed alveolar proteinosis. A bone marrow rejection or a depopulation of the lungs from donor alveolar macrophages was suspected.
DM, AF, SB and BN: However, chimerisms on peripheral whole blood and BAL were 100% donor. We suspected an adverse side effect of rapamycin and suggested discontinuing this treatment. The patient slowly recovered over 9 months, allowing progressive tapering and cessation of oxygen therapy (figure 1G).
AH, CS, CD and JDB: Following severe acute GVHD after HSCT and this episode of severe respiratory impairment, successive pulmonary function tests displayed non-reversible obstructive lung disease, gas trapping associated with normal gas exchange suggestive of obliterative bronchiolitis (OB).
CoB, GC, AH, CP and MLF: Systemic immunosuppression was stopped 26 months after HSCT (figure 1G). At last follow-up, 40 months after HSCT, the patient suffers from ophthalmic chronic GVHD requiring topical immunosuppressive treatment; she has a normal respiratory examination and her exercise capacity continues to improve as confirmed by a normal 6 min walking test. Pulmonary function test results remained stable, suggesting that the OB had not progressed.
MLF, DM, AF, SB, CS, Marina Cavazzana (MC), CD, JBD and BN: Although our patient was suffering from severe PAP prior to HSCT, the rapid, complete resolution of respiratory symptoms was suggestive that the full recovery of donor-derived haematopoietic function led to alveolar macrophage replacement. Rapid colonisation of the lungs by donor alveolar macrophage was indeed documented by a BAL performed on D15 post-HSCT (ie, 94% of cells in the BAL fluid were XY). Furthermore, the 6 months’ post-transplantation CT scan was normal. This unique observation proves that bone marrow reconstitution from a healthy donor provides alveolar macrophages expressing GM-CSF-R, and enables effective surfactant homeostasis. Although this approach was successful in a murine model of hereditary PAP (in csf2rb-/- mice),3 the present case is (to the best of our knowledge) the first successful HSCT in humans for the treatment of PAP. Genoidentical HSCT was attempted in a 3-year-old girl with GM-CSF-Rα deficiency but the patient died of a respiratory infection before haematological recovery. Despite these promising results, HSCT remains a complex procedure with relatively high morbidity and mortality rates—especially in compromised patients. In the future, alternative treatment options might also be considered. Haematopoietic stem cell gene transfer appears to be promising in this condition, as shown by experiments in the csf2rb-/- mouse model. Indeed, transplantation of haematopoietic stem cells transfected with a gamma retroviral vector expressing the murine csf2rb gene in lethally irradiated csf2rb-/- mice resolved the PAP, despite a low level of transduction.4 This approach minimises transplant-related mortality (by avoiding GVHD and the need for immunosuppression) but still requires myeloablation. Organ-targeted, cell-specific therapy with pulmonary macrophage transplantation is a promising procedure in csf2rb-/- mice. In the absence of any myeloablation, a single instillation of wild type (WT) or csf2rb-/- gene-corrected, bone-marrow-derived macrophages was able to correct the lung disease and normalise disease-specific biomarkers for at least 1 year. Importantly, this model highlighted the WT cells’ survival advantage over csf2rb-/- cells—a property of primary importance for successful cell therapy.5
MLF and BN: In conclusion, we report on the first successful curative treatment of congenital PAP caused by GM-CSF-R deficiency in humans. Our observation confirms that HSCT is a potentially valuable treatment approach but is also associated with a high risk of infection, GVHD and drug toxicity. This case supports the feasibility of translational clinical studies of cell therapies tested in animal models of PAP.
Acknowledgments
The authors thank the patient and her family.
Footnotes
Contributors MLF, AH, CS, SB, AF and BN contributed to the manuscript preparation. LB contributed to the imaging data analysis. JB contributed to the histopathological analysis. All authors critically read, commented on, and approved the final version of the manuscript.
Funding Institut National de la Santé et de la Recherche Médicale (grant number 000427993).
Competing interests None declared.
Patient consent Guardian consent obtained.
Ethics approval Comité de Protection des Personnes (CPP).
Provenance and peer review Not commissioned; externally peer reviewed.