Article Text

Statistics from Altmetric.com
As spring emerges from the shadows of a cold, dark winter, we are again left to reflect on the ravages of another influenza season. Based on the emergent data from Australia,1 the 2017–2018 outbreak in the Northern Hemisphere was predicted to be particularly severe and so it proved to be. Disease severity can be driven by a number of factors, including shifts in the viral genome, but host innate immune responses also play a role, most notably in the lethal cytokine storm response to avian influenza strains.2
Macrophages are the predominant innate immune cell in the human airway and critical for an appropriate protective response to bacterial, fungal and viral infections. These cells enhance viral clearance, being essential for both viral antigen presentation to mucosal T cells3 and the development of neutralising antibodies.4 Furthermore, when macrophages are depleted, there is increased lung pathology5 in response to viral infection demonstrating the central role of macrophages in ensuring that virus control does not compromise host survival. In the majority of cases, such mechanisms ensure that lung inflammation is kept to a minimum so that pneumonias do not develop. However, it is known that influenza infection can lead to both primary and secondary bacterial pneumonias,6 although whether as a result of virus or host factors is unclear. Pneumonias are primarily associated with massive neutrophil influx into the lung leading to compromised gas exchange but there are conflicting reports as to whether the role of the neutrophil in viral infections is protective or pathological.7 The study by Peiró et al8 has provided further insight into the complex interplay between macrophages and neutrophils in a mouse model of influenza infection.
Peiró and colleagues first demonstrated that the pleiotropic proinflammatory cytokine, interleukin 1-beta (IL-1β), is induced by influenza infection and that the amount of this cytokine released correlates with neutrophil infiltration into the infected lung. IL-1β is an acute phase cytokine that once released binds the IL-1 receptor, causing further release of IL-1β and other inflammatory cytokines and chemokines. Activation of viral pattern recognition receptors, such as Toll-like receptor (TLR)3 and 7 or retinoic acid-inducible gene I, activate IL-1β gene expression via the transcription factor NF-κB (nuclear factor-κB) (figure 1). Because of the potent nature of IL-1β, the mRNA generated is translated into a proform of the protein that requires further processing to produce mature IL-1β by the enzyme caspase-1.9 Caspase-1 is activated by the nucleotide-binding and oligomerisation domain-like receptor protein (NLRP)-3 inflammasome which can be activated by other damage-associated molecular patterns (DAMPs) such as ATP.
{kind=link}
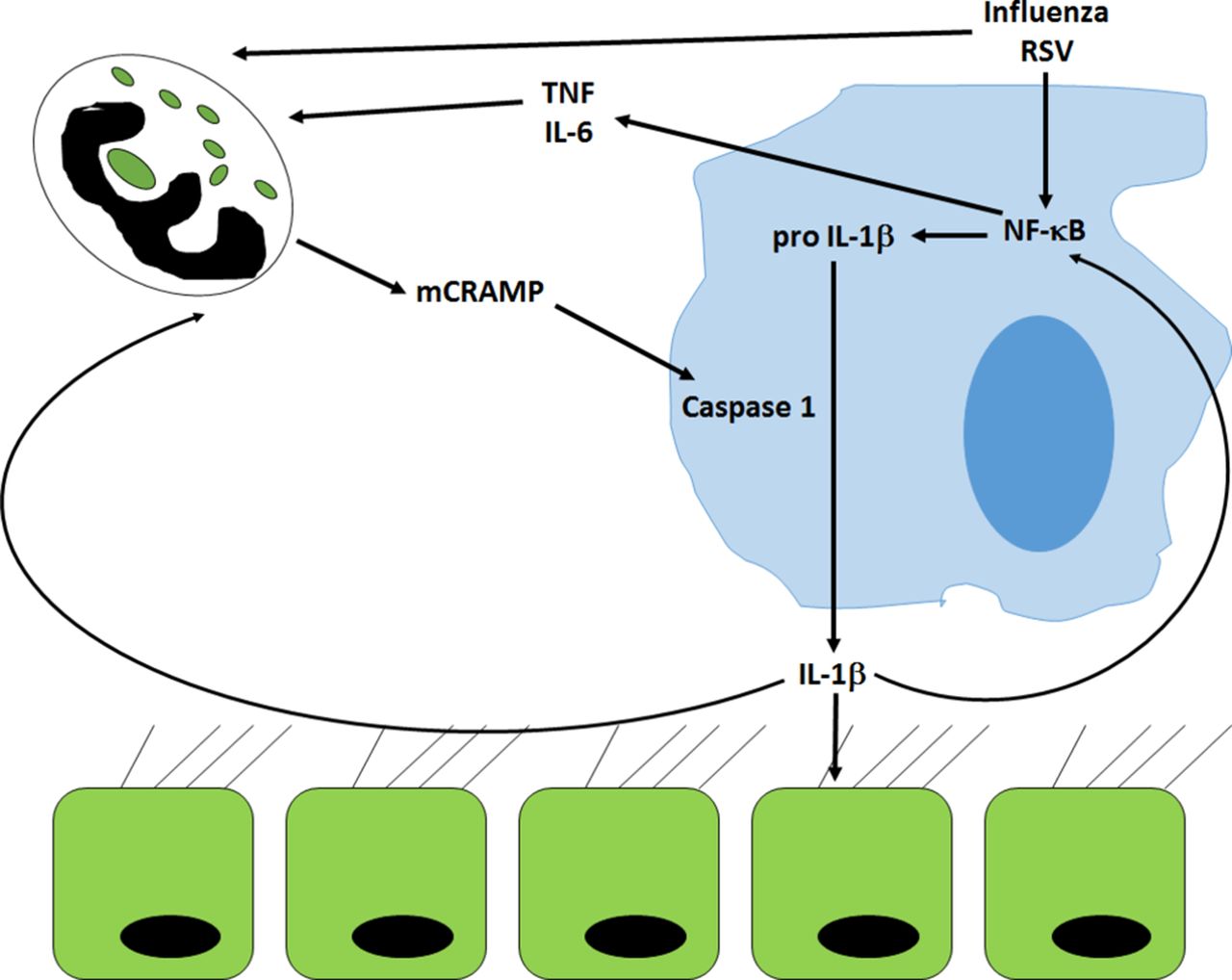
Mediation of cross-talk between alveolar macrophages and neutrophils by mCRAMP. Infection by viruses causes stimulates proinflammatory pathways that lead to activation of NF-κB in macrophages. This activation leads to the accumulation of pro-IL-1β, which can only be cleaved to its active form by caspase-1. Caspase-1 is activated by the murine LL-37 orthologue, mCRAMP, released from activated neutrophils. The subsequent release of active IL-1β acts in a paracrine fashion on both neutrophils and epithelial cells and feeds back onto macrophages stimulating further downstream proinflammatory pathways. The activation of these pathways may be responsible for the development of ALI/ARDS in response to viral infection. ALI/ARDS, acute lung injury/acute respiratory distress syndrome; IL-1β, interleukin 1-beta; mCRAMP, murine cathelicidin -related antimicrobial peptide; NF-κB, nuclear factor-κB; RSV, respiratory syncytial virus; TNF, tumour necrosis factor.
Antibody-mediated depletion of neutrophils specifically reduced the amount of influenza-induced IL-1β but not IL-6 or tumour necrosis factor-alpha, suggesting that the neutrophils were directly responsible for this increase in IL-1β.8 Moreover, neutrophil depletion only reduced viral-induced, not bacterial-induced, release of IL-1β. However, on further investigation by both immunohistochemistry and flow cytometry, it was the macrophages that were positive for IL-1β in response to influenza infection, not the neutrophils. As the antibody used could not distinguish between the pro and active form of IL-1β, there was no obvious effect of neutrophil depletion on the expression of IL-1β by macrophages. That was until the study team probed activation of caspase-1 in influenza-infected lung, which was markedly reduced on neutrophil depletion. To deconvolute what signal could be coming from the neutrophils that would activate the NLRP3 inflammasome and cause release of mature IL-1β from macrophages, Peiró and colleagues first investigated the DAMPs, ATP and uric acid. The release of neither of these compounds was affected by neutrophil depletion in bronchoalveolar lavage fluid from influenza-infected animals. Similarly, administration of compounds that inhibit these two DAMPs were also found to be ineffective. In contrast, incubating the cells in the presence of the murine homologue of the human cathelicidin, LL-37 (mCRAMP) led to a profound increase of the amount of IL-1β released from alveolar macrophages treated with the TLR7 agonist, imiquimod. Furthermore, detection of mCRAMP was substantially reduced in neutrophil-depleted, influenza-infected animals and the amount of IL-1β released correlated with the amount of mCRAMP detected.
The cathelicidin LL-37 is an amphipathic protein that has direct antimicrobial and antiviral effects as well as being a chemoattractant for neutrophils, monocytes and T cells.10 In addition to directly activating the NLRP3 inflammasome, LL-37 can also enhance the ability of neutrophils to form neutrophil extracellular traps (NETs) in response to influenza,11 an effect that is also promoted by IL-1β.12 Thus, it could be envisaged that the neutrophils are recruited to the influenza-infected lung to try and contain the virus by releasing NETs. Once trapped, the virus could then be killed by the effects of the LL-37 among other mediators, before being phagocytosed and cleared by activated macrophages.
Unfortunately, this is too neat a story and is one that is not supported by Peiró et al’s study. Depletion of neutrophils had no effect on the amount of influenza or respiratory syncytial virus (RSV) shed in this murine model. Furthermore, the small reduction in symptoms reported was only seen in the neutrophil-depleted group suggesting that the neutrophils were driving symptoms while having little effect on viral clearance. Thus, the Peiró et al study appears to give further weight to the pathological role of neutrophils in influenza infection by removing a critical brake on macrophage cytokine production. The IL-1β released from the activated macrophages can act in an autocrine fashion13 as well as in a paracrine positive feedback loop onto the lung epithelial cells14 causing further inflammatory cytokine release (figure 1). When combined with further neutrophil activation and cytokine release, this may explain the cytokine storm and acute lung injury/acute respiratory distress syndrome associated with the most severe influenza infections.
The major limitation of this work is the lack of any human data to support the murine model. Mouse neutrophils are known to differ from those of human cells in terms of their granule contents and also their response to stimulation (reviewed in15). In addition, the authors were unable to show whether the macrophages were themselves infected by virus, as has been shown in human tissue,6 or whether the macrophages were reacting to epithelial infection. Furthermore, there is no one gold-standard murine model of influenza infection, with different strains and titres of influenza being administered via different routes in mice of different genetic backgrounds. Such differences in models may explain some of the contradictory reports in the literature. Despite these caveats, the strength of murine modelling is in the ability to investigate the response of the whole organism to viral challenge. Intriguingly, Peiró et al demonstrate that RSV recruits neutrophils and releases IL-1β in a similar manner to influenza, suggesting that these are common responses to infection by RNA viruses.
The questions that remain are if neutrophils are so detrimental in influenza infection, what factors are responsible for the recruitment of neutrophils to the airway in response to this virus and why are they recruited in the first place? Severe infections are often associated with chronic lung diseases, like severe asthma and chronic obstructive pulmonary disease (COPD), both of which are not only associated with neutrophilia and increased LL-3716 17 but also a dysbiosis of the lung microbiome,18 with increased abundance of Proteobacteria spp and non-typeable Haemophilus influenzae (NTHi) in particular. IL-1β is already increased in the lungs of patients with COPD who are colonised with NTHi,19 and compellingly in a longitudinal cohort of patients with COPD, we observed that NTHi colonisation increased the risk of a viral infection being associated with an exacerbation compared with the presence of virus alone.20 NTHi is also frequently detected in the sputum of hospitalised patients with pneumonia.21 As Peiró and colleagues demonstrate the differential effects of viruses and bacteria on inflammasome activation, it could indicate that the macrophages in bacterially colonised patients are already on a hair trigger, with virus infection then being enough to initiate a run-away inflammatory response. For now this scenario must remain speculation awaiting future work, but in the meantime Peiró and colleagues have given us a good insight into what these initiating factors might be. What is now required is further understanding of how these mechanisms are regulated, so that this inflammatory chain reaction can be switched off. This off-switch needs to be applied after pathogen clearance but, most importantly, before development of pneumonia allowing macrophages and neutrophils to be part of the (re)solution rather than part of the problem.
References
Footnotes
Contributors KJS is the sole contributor.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.