Article Text

Abstract
Background Neutrophils may cause tissue disruption during migration and by releasing cytotoxic molecules. However, the benefits of neutrophil depletion observed in experimental models of lung injury do not correspond with the poor outcome of neutropenic patients.
Methods To clarify the role of neutrophils during repair, mice with ventilator induced lung injury (VILI) were rendered neutropenic after damage, and followed for 48 hours of spontaneous breathing. Lungs were harvested and inflammatory mediators and matrix metalloproteinases measured. Bronchoalveolar lavage fluid (BALF) from ventilated patients with acute respiratory distress syndrome, with or without neutropenia, was collected, the same mediators measured and their effects in an ex vivo model of alveolar repair studied. Finally, neutropenic mice were treated after VILI with exogenous matrix metalloproteinase-9 (MMP-9).
Results Lungs from neutropenic animals showed delayed repair and displayed higher levels of tumour necrosis factor α, interferon γ and macrophage inflammatory protein 2, and absence of MMP-9. BALF from ventilated neutropenic patients with acute respiratory distress syndrome showed similar results. BALFs from neutropenic patients yielded a delayed closure rate of epithelial wounds ex vivo, which was improved by removal of collagen or addition of exogenous MMP-9. Lastly, treatment of neutropenic mice with exogenous MMP-9 after VILI reduced tissue damage without modifying cytokine concentrations.
Conclusion Release of MMP-9 from neutrophils is required for adequate matrix processing and lung repair.
- ARDS
- immunodeficiency
- lung proteases
- Neutrophil Biology
Statistics from Altmetric.com
Key messages
What is the key question?
Why do neutropenic patients exhibit such a poor outcome when undergoing mechanical ventilation?
What is the bottom line?
Using in vivo and ex vivo models with neutropenic mice and samples from neutropenic patients, respectively, we shown impaired repair after ventilator induced lung injury, related to a deficiency in alveolar matrix metalloproteinase-9.
Why read on?
Our findings can explain the reasons behind the poor prognosis of neutropenic patients under mechanical ventilation and the negative impact of anti-inflammatory therapies in the acutely injured lung, while opening a new therapeutic approach.
Introduction
Neutrophils are myeloid cells recruited to sites of injury at the early phases of the inflammatory response. Once activated, they release a wide variety of molecules aimed at eradicating the pathogenic stimulus. These molecules include proinflammatory cytokines, proteases and free radicals. During this initial response, the host tissue can be injured even further due to the action of neutrophils.1
Due to their contribution to tissue damage in the early stages of the inflammatory response, neutrophil depletion or their functional blockade using antagonists of chemotaxis have demonstrated substantial benefits in experimental models of acute critical illness, including lipopolysaccharide injection,2 3 complement activation4 or transfusion related acute lung injury.5 Similarly, lung injury caused by mechanical ventilation with high pressures and volumes (so called ventilator induced lung injury (VILI)) is promoted by neutrophils, and its depletion6 or inhibition7 decreases tissue injury. These models have shown that neutrophilic infiltration can be a significant part of the pathogenic response during inflammation. Accordingly, strategies aimed at interfering with neutrophils have been proposed in acute inflammatory syndromes, such as sepsis or acute respiratory distress syndrome (ARDS).8
These experimental findings, however, contrast with the clinical outcomes in neutropenic patients. This population, especially in the setting of haematological diseases, frequently develops lung injury and shows mortality rates up to 80% if mechanical ventilation is needed.9 There are several explanations for the discrepancy between the benefits of experimental neutropenia and its clinical consequences. Among these, the presence of an active infection is commonly considered the most relevant. However, the majority of neutropenic patients receive antibiotic treatments early on, and non-infectious conditions or absence of positive cultures in this setting have been related to even worse outcomes.10 In addition, most neutropenic patients have low counts of other immune cells, such as monocytes, that may be involved in lung damage.11
We have therefore proposed a novel, additional explanation: neutrophils may in fact act as a double edged sword, promoting lung injury but also contributing to tissue repair. Neutrophils are a source of proteolytic enzymes that can cleave inflammatory mediators, leading to their inactivation, and process the newly synthetised collagen, avoiding the formation of a scar and promoting tissue repair. In the specific case of VILI, we observed that the persistence of neutrophils during the repair phase, and the subsequent increase in gelatinases matrix metalloproteinase-9 (MMP-9) and matrix metalloproteinase-2 (MMP-2), was related to a better outcome in an experimental model of lung repair.12 Therefore, we hypothesised that neutrophils may play a critical role during the subsequent repair phase of VILI. To test this without interfering with the initial pathogenic role of neutrophils, we induced neutropenia after the onset of injury, and studied the repair phase and the molecular mechanisms involved.
Methods
A complete description of the methods can be found in the online supplementary file.
supplementary file
Experimental model
The Animal Research Ethics Committee of the Universidad de Oviedo evaluated and approved the study.
Mice were anaesthetised, intubated and ventilated in pressure controlled mode (peak inspiratory pressure 17 cm H2O; positive end-expiratory pressure 0 cm H2O; respiratory rate 50 breaths/min; I:E ratio 1:1; inspired oxygen fraction 0.21). After 90 min of ventilation, mice were recovered from anaesthesia, extubated and allowed to breath spontaneously for 2 days. During the recovery phase, animals were randomised to receive an antineutrophil antibody to induce neutropenia or the corresponding isotype control antibody (see below). Induction of neutropenia was confirmed by peripheral blood counts. Overall, the experimental setup thus included four groups for analysis of lung injury (baseline, VILI, repair, repair+neutropenia), with 17 animals per group (figure 1A). Ten of these animals were used for tissue sampling. In three animals per group, the right lungs were weighed before and after drying in an oven at 55°C for 72 hours to calculate the wet to dry lung weight ratios. MicroCT scans of the left lungs of these animals were acquired in a SkyScan-1174 scanner (see online supplementary file for details). In the four remaining animals, a bronchoalveolar lavage was performed, and the albumin concentration measured in the obtained bronchoalveolar lavage fluid (BALF).

Lung injury and repair in each experimental group. (A) The experimental design and representative histological sections. Mice were studied under baseline conditions, immediately after ventilator induced lung injury (VILI) and after 48 hours of repair. Animals were randomised to receive an anti-Gr1 antibody or unspecific IgG during this repair phase. Induction of neutropenia was related to a more severe lung damage after 48 hours of repair (B) (p=0.004, ANOVA). There were no differences in wet to dry weight ratios (C) (p=0.12, ANOVA) or albumin concentration in bronchoalveolar lavage fluid (D) (p=0.08, ANOVA). The extent of injury was evaluated using CT scans. The fraction of the non-aerated lung volume (defined as a density >−100 Hounsfield units) was significantly higher immediately after injury and in neutropenic mice after 48 hours of spontaneous ventilation compared with healthy controls (E) (p<0.01, ANOVA). Representative volumetric renderings (identifying normally aerated and non-aerated lung in blue and yellow–red respectively), CT slices and average density histograms (F). FiO2, inspired oxygen fraction; PIP, peak inspiratory pressure; PEEP, positive end-expiratory pressure.
In separate experiments, neutropenic animals were randomly assigned to receive MMP-9 (six intranasal doses of 25 ng/8 hours, n=9) or the equivalent volume of saline (10 μL, n=9) after induction of VILI.
Tissue sampling
Lungs were removed from anaesthetised mice as previously described.13 Histological studies were performed in paraffin embedded tissues, using a predefined score ranging from 0 (normal lung) to 4 (massive disruption of lung architecture).14 The number of neutrophils, macrophages and Ki67 positive cells were counted in three random fields in immunostained sections (with anti-myeloperoxidase, F4/80 and anti-KI67 antibodies, respectively). All analyses were performed by two observers blinded to the experimental conditions.
Patient lavage samples
Mini-bronchoalveolar lavages were performed as described15 in four non-neutropenic and four neutropenic patients under mechanical ventilation during the early phase of ARDS (<3 days from meeting ARDS criteria). Clinical data of these patients are shown in the online supplementary table 1. This part of the protocol was reviewed and approved by the regional ethics committee (Comité Ético de Investigación Clínica del Principado de Asturias, Spain). Signed informed consent was obtained from each patient’s next of kin.
Biochemical measurements
Levels of tumour necrosis factor α (TNFα), interferon γ (IFNγ), macrophage inflammatory protein 2 (MIP-2) (or interleukin 8 (IL-8) in human samples), IL-6, IL-10, MMP-2, MMP-8, MMP-9 and tissue inhibitor of metalloproteinase-1 (TIMP-1) were measured.15 16 Additionally, expression of Myc and cyclin-D1 (Ccnd1) were assessed by quantitative PCR.
Wound healing assay
To characterise the potential of BALF from patients to induce lung repair, wound closure was studied in monolayers of BEAS-2b cultured in the presence of these fluids. The assays were performed in plates with and without a type I collagen coating (Oris Cell Migration Assays, AMS Biotechnology, UK). In separate experiments, exogenous MMP-9 was added to the BALF in collagen coated plates.
Statistical analysis
Data are presented as mean±SEM. T tests, ANOVA or repeated measure ANOVA were used for bivariate and multiple comparisons, respectively (see online supplementary file for details). When appropriate, post hoc comparisons were done using Holm’s correction. A p value <0.05 was considered significant. All calculations were done using the R 3.2.1 statistical package.
Results
Neutropenia after VILI impairs repair
To clarify the role of neutrophils during repair after VILI, neutropenia was induced after high pressure ventilation by treatment with anti-Gr1 antibodies. Treated mice showed a rapid decrease in neutrophil counts in peripheral blood, while these remained constant in animals receiving the isotype control (0.59±0.76 vs 3.33±1.39×109 neutrophils/L, 24 hours after extubation). After 48 hours of spontaneous breathing, histological injury scores were significantly decreased in isotype control treated mice compared with values obtained immediately after VILI. Neutropenic animals, however, did not show a significant change in lung damage, indicating an inability to resolve the initial injury (figure 1B). Both wet to dry weight ratio (figure 1C) and albumin (figure 1D) concentration in BALF increased after VILI and decreased in 48 hours. However, small changes and the large variability precluded any statistically significant result. Lung CT scans showed an increase in non-aerated areas after VILI. These non-aerated volumes decreased after 48 hours in vehicle treated but not in neutropenic mice (figure 1E). Figure 1F shows volumetric reconstructions of representative CT scans, a CT scan slice and density histograms of each experimental group.
Neutrophilic infiltrates within the lung increased immediately after VILI and were decreased 48 hours later. Mice treated with the anti-Gr1 antibody showed less neutrophils within the lung parenchyma (figure 2A). The macrophage count increased after VILI (figure 2B) and decreased during repair, with no differences between neutropenic and control mice. Cell proliferation, assessed by Ki67 immunostaining, increased after VILI and decreased in the repair phase (figure 2C). Figure 2D shows representative histological sections of these studies.

Cell populations. Regarding immune cells, neutrophils (A) (p<0.001, ANOVA) and macrophages (B) (p=0.003, ANOVA) were counted (myeloperoxidase (MPO) and F4/80 positive cells, respectively) in histological sections. Proliferating cells were identified by Ki67 staining. (C) (p=0.002, ANOVA). (D) Representative sections of each experimental group. Significant differences in post hoc tests are shown on the graphs. VILI, ventilator induced lung injury.
Inflammatory mediators are increased in lungs from neutropenic mice
The abundance of pro- and anti-inflammatory cytokines was studied in lung tissue from neutropenic and control animals. Interestingly, lungs from neutropenic mice showed higher levels in the majority of proinflammatory cytokines studied, including TNFα (figure 3A), IFNγ (figure 3B) and MIP-2 (figure 3C). However, there were no differences in IL-6 (figure 3D) or IL-10 (figure 3E).

Inflammatory mediators and matrix metalloproteinases during the repair phase (48 hours after ventilation). Abundance of tumour necrosis factor α (TNFα) (A), interferon γ (IFNγ) (B), macrophage inflammatory protein 2 (MIP-2) (C), interleukin 6 (IL-6) (D) and interleukin 10 (IL-10) (E) was measured in lung homogenates by western blotting. Lungs from neutropenic mice showed increased levels of the proinflammatory mediators TNFα (p=0.04), IFNγ (p=0.04) and MIP-2 (p=0.03). Levels of matrix metalloproteinase-8 (MMP-8) (F) (p=0.08), MMP-2 (G) (p=0.70) and MMP-9 (H–I), corresponding to pro-MMP-9 and active MMP-9 (p<0.01 and p=0.05, respectively) and the tissue inhibitor of MMP-1 (TIMP-1) (J) (p=0.59) were measured in lung homogenates. There was a significant decrease in MMP-9 in lungs from neutropenic animals, with no differences in the other molecules. (K) Representative western blots and a zymography. *p<0.05 compared with non-neutropenic counterparts. VILI, ventilator induced lung injury.
As matrix remodelling poses a key mechanism of tissue injury and repair, we next probed for protein levels of MMP-8, MMP-2, MMP-9 and TIMP-1. There were no significant differences in levels of MMP-8 (figure 3F) or total MMP-2 (figure 3G). Yet, both the inactivated and activated MMP-9 forms were significantly decreased in neutropenic mice (figure 3H-I). As the TIMP-1 levels were not modified by neutropenia (figure 3J), this finding would suggest decreased MMP-9 activity after neutrophil depletion. figure 3K shows representative western blots and a zymography.
It has been reported that neutrophil migration across the alveolar epithelium may activate the Wnt pathway, thus promoting tissue repair. To address this issue, we quantified Myc and Ccnd1 expression in samples from control and neutropenic mice. There were no detectable differences in any of these genes (online supplementary figure 1).
Inflammatory mediators are increased in BALF from neutropenic patients
To test for the validity of our findings for human disease, we studied cytokine and MMP levels in BALF from neutropenic and non-neutropenic patients who underwent mechanical ventilation. Clinical data of these patients are shown in the online supplementary table 1. BALF from neutropenic patients showed higher levels of TNFα (figure 4A), IFNγ (figure 4B) and IL-8 (figure 4C). As in the animal model, there were no significant differences in IL-6 (figure 4D) and IL-10 (figure 4E). Regarding MMPs, there were no differences in MMP-8 (figure 4F) and MMP-2 (figure 4G). Again, both pro- and active MMP-9 were significantly lower in BALF from neutropenic patients (figure 4H and I, respectively). Concentrations of TIMP-1 (figure 4J) were similar between groups. Figure 4K shows a representative immunoblot for MMP-8 and a zymography of patient samples. These results support the validity of our experimental model and highlight the differential immune response during mechanical ventilation and neutropenia.

Inflammatory mediators and matrix metalloproteinases (MMPs) in neutropenic patients. Concentrations of the cyto/chemokines tumour necrosis factor α (TNFα) (A) (p=0.05), interferon γ (IFNγ) (B) (p=0.04), interleukin 8 (IL-8) (C) (p=0.04), interleukin 6 (IL-6) (D) (p=0.65), interleukin 10 (IL-10) (E) (p=0.69), MMP-8 (F) (p=0.24), MMP-2 (G) (p=0.72), pro-MMP-9 (H) (p<0.01), active MMP-9 (I) (p=0.03) and tissue inhibitor of metalloproteinase-1 (TIMP-1) (J) (p=0.86) were measured in bronchoalveolar lavage fluid from neutropenic and non-neutropenic patients with acute respiratory distress syndrome and mechanical ventilation. Similar to the animal model, neutropenic patients showed higher levels of proinflammatory cytokines and a significant decrease in MMP-9. (K) Representative western blot and zymography of these samples. *p<0.05 compared with non-neutropenic counterparts.
Delayed alveolar wound closure during neutropenia depends on matrix processing
Next, we studied the potential of BALF from neutropenic and control patients to promote epithelial repair. To this end, we measured the wound closure rates of BEAS-2b cells cultured on a type I collagen matrix in the presence of these BALFs. Globally, closure rates were always accelerated in the presence of BALF compared with culture medium alone (upper limit of the grey areas in figure 5), illustrating its ability to promote epithelial repair. However, as shown in figure 5A, wound closure was delayed when BALF from neutropenic patients was added to the culture medium. To characterise the role of the extracellular matrix in these differences, we repeated the experiments in two different conditions. When cells were seeded onto plates without collagen, the differences changed, and BALF from neutropenic patients accelerated wound closure (figure 5B). In contrast, addition of exogenous recombinant human MMP-9 to BALFs abolished the differences between samples from neutropenic and non-neutropenic patients, rendering closure curves almost identical (figure 5C). Moreover, these two curves were also identical to the closure rate observed in presence of fetal bovine serum, suggesting MMP-9 as a critical factor that mediates BALF induced acceleration of wound closure. Differences in closure rates were also evaluated by calculation of the half-lives (figure 5D). Collectively, these results demonstrate that the differences in wound closure between neutropenic and control patients are related to differential collagen processing, with MMP-9 playing a central role.

Wound healing in presence of bronchoalveolar lavage fluid (BALF) from neutropenic and non-neutropenic patients. Wound closure rates of BEAS-2b cell monolayers were assessed in culture plates with (A) and without (B) collagen coating. BALF from neutropenic patients delayed closure in the presence of collagen (A) (p=0.003, repeated measures ANOVA) and accelerated closure in plates without collagen (B) (p<0.001, repeated measures ANOVA). Addition of exogenous matrix metalloproteinase-9 (MMP-9) to the culture medium equaled the closure rates between BALFs from both groups (C) (p=0.87). The grey areas depict the closure in the presence of fetal bovine serum (fastest closure) and without any additive other than culture medium (slowest). Closure half-lives (t1/2) are shown in (D). *p<0.05 compared with non-neutropenic counterparts.
Exogenous MMP-9 improves repair in neutropenic mice
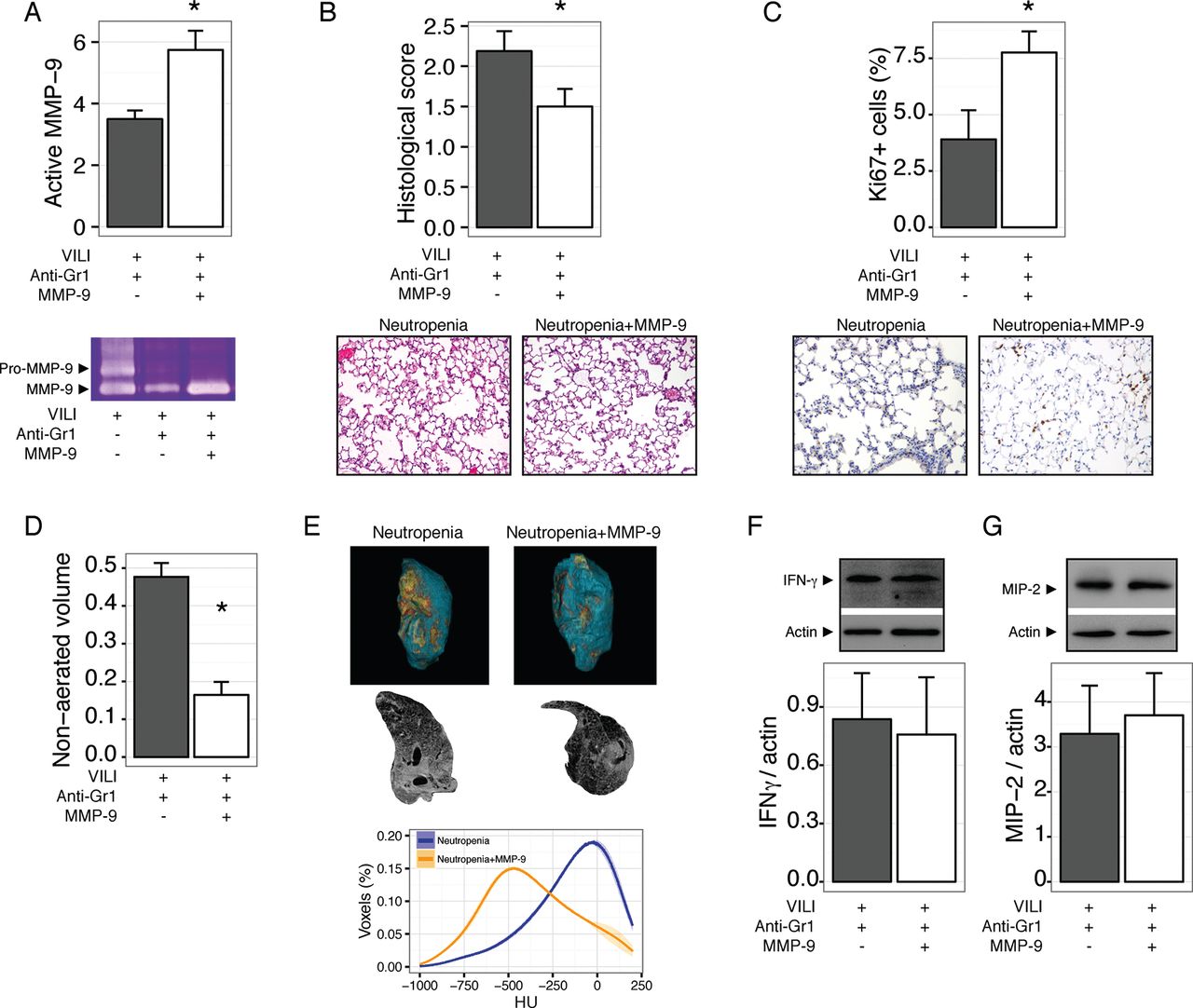
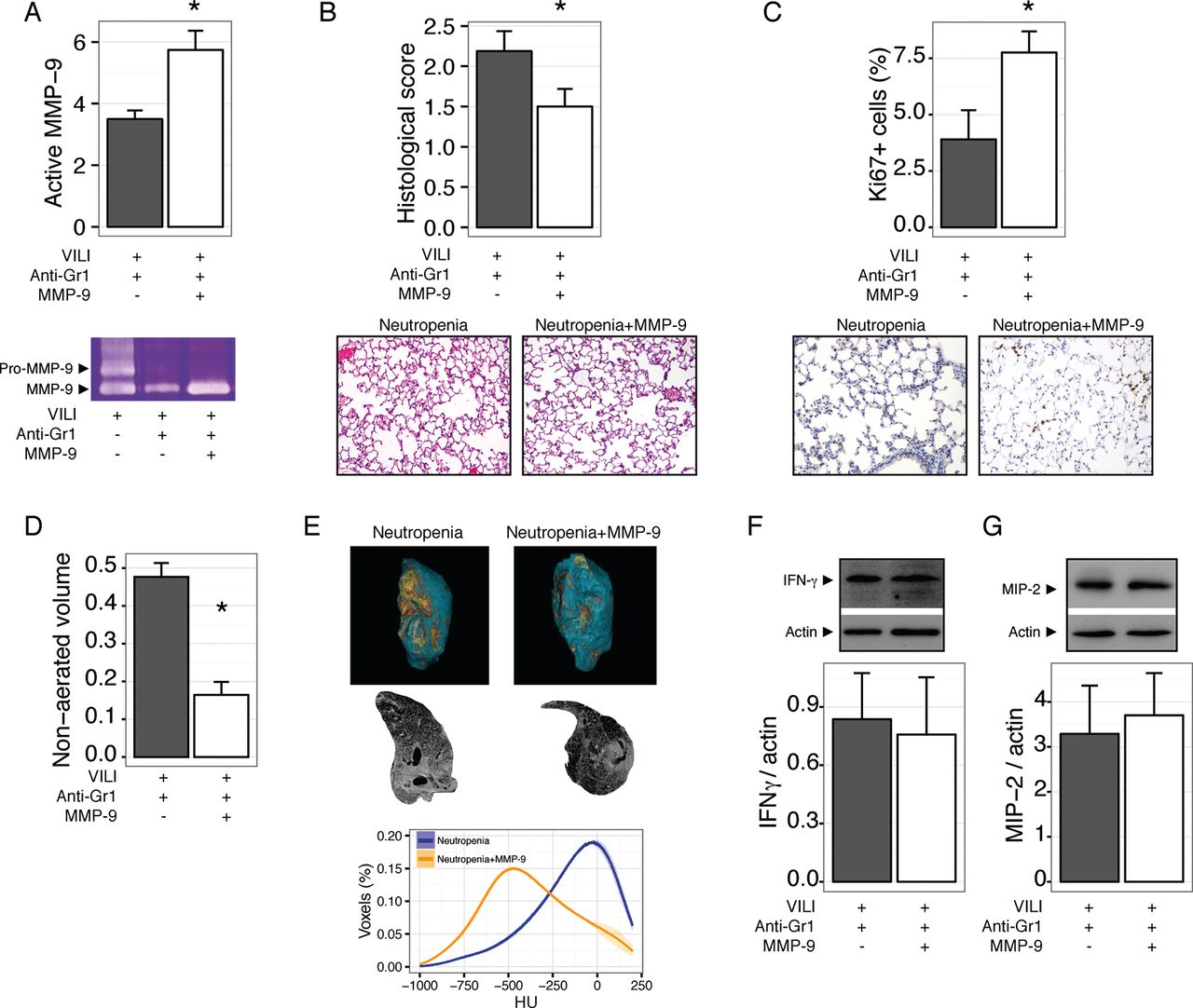
Our results so far show that neutropenic animals have impaired repair after VILI, and that this could be related to abnormal processing of the extracellular matrix due to the decreased abundance of MMP-9 in neutropenic BALF. To test this notion in an in vivo setting, we treated mice with neutropenia after VILI with inhaled MMP-9. Exogenous MMP-9 increased the levels of this gelatinase in lung homogenates (figure 6A). After 48 hours, MMP-9 treated mice showed improved tissue repair, as shown in figure 6B, with a significant increase in proliferating cells (figure 6C). MMP-9 treatment did not modify the wet to dry weight ratio (4.0±0.22 and 3.8±0.04 for vehicle and MMP-9 treated neutropenic mice, respectively, p=0.2). Decreased lung damage in MMP-9 treated mice was also reflected in lower non-aerated lung volumes in CT scans (figure 6D-E). Interestingly, exogenous MMP-9 did not decrease the levels of proinflammatory cytokines IFNγ (figure 6F) and MIP-2 (figure 6G).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Exogenous matrix metalloproteinase-9 (MMP-9) improves lung repair in neutropenic mice. Mice were rendered neutropenic by anti-Gr1 administration after ventilator induced lung injury (VILI) and treated with inhaled MMP-9 or vehicle. Treatment with the enzyme increased its concentration in lung homogenates (A) and decreased lung damage after 48 hours (B). Proliferating cells, as measured by Ki67 staining, were increased after MMP-9 treatment (C). In line with these findings, the fraction of non-aerated lung volume also decreased after MMP-9 treatment (D, E). Levels of interferon γ (IFNγ) (F) and macrophage inflammatory protein 2 (MIP-2) (G) in lungs homogenates were not modified by the treatment. *p<0.05 compared with vehicle treated animals.
Discussion
The main findings of our work are that neutropenia causes upregulation of the immune response during the repair phase of VILI, delayed tissue repair and a significant reduction in MMP-9. Data from patients with ARDS confirmed the increase in proinflammatory mediators and absence of MMP-9 during neutropenia observed in the animal model. Reconstitution of MMP-9 both in vivo and in vitro improves lung and epithelial repair, respectively. Collectively, our findings support a key role for neutrophil derived MMP-9 in the epithelial repair phase after tissue damage, and provide an additional explanation for both the poor outcome of neutropenic patients undergoing mechanical ventilation and the lack of benefits of anti-inflammatory therapies in ARDS.
Neutrophil recruitment from the circulation to sites of injury is frequently associated with loss of endothelial or epithelial integrity.17 Release of cytotoxic and proteolytic molecules from their granules may cause or aggravate tissue injury.18 Therefore, neutrophils have been implicated in organ injury in a large variety of different diseases, including ARDS. In line with their pathogenic role, experimental models of lung injury have shown that neutrophil depletion6 or blockade of chemotactic receptors may have a protective role.7 13 In the special case of VILI, the neutrophilic infiltrate is one of the first steps towards lung damage, and its inhibition markedly reduces injury.7 By these findings, targeting neutrophils has been proposed as a therapeutic goal to prevent ARDS.19
These data contrast with the poor outcome of neutropenic, critically ill patients. In this population, acute lung injury is common, and results in a high mortality rate if mechanical ventilation is needed.20 Previous hypotheses have attributed this contradiction to the inability of neutropenic patients to clear their lungs from invading pathogens, to activation of other injurious mechanisms by the underlying disease or to the ability of neutrophils to cause lung injury even at relatively low numbers. We illustrate here an additional explanation in that neutrophils also play a role in the subsequent resolution of lung injury. We previously showed that persistence of neutrophils during the repair phase was related to better survival.12 These results and the findings reported herein suggest that the main differences between neutropenic and non-neutropenic animals are due to changes in lung structure (cell proliferation and matrix remodelling) rather than oedema clearance (due to the absence of changes in wet to dry weight ratio or albumin content). The release of proteolytic enzymes from neutrophils causes activation of the Wnt/β-catenin pathway, thus activating tissue repair.17 However, the absence of changes in cyclin-D1 (Ccnd1) and Myc expression suggests that this route is not differentially activated in our experimental model. An additional study focused on acid induced lung injury found increased damage in neutropenic mice, and proteomic profiling has identified several matrix metalloproteinases as putative mediators responsible for the differences with non-neutropenic animals.21
Matrix metalloproteinases can cause tissue disruption and modulate the inflammatory response by the cleavage of mediators,22 23 but may also contribute to tissue remodelling and repair.24 We previously demonstrated that MMP-2 and MMP-9 are protective in VILI.12 25 Analogously, MMP-9 has been related to a better outcome in ARDS patients.26 In line with these findings, the present work shows that absence of neutrophil derived MMP-9 delays migration of lung epithelial cells, and that its supplementation can restore epithelial wound closure in vitro, and revert the phenotype of neutropenic animals in vivo. The small, although significant, differences in cell culture experiments contrast with the large differences in in vivo models. This could be due to the arbitrary doses of BALF used in the cultures, aimed at illustrating the operating mechanisms rather than reflecting the real alveolar conditions.
We also identified a preponderance of inflammatory cytokines, including TNFα, IFNγ and MIP-2, in neutropenic compared with non-neutropenic mice. Analyses of BALF from patients confirm these differences. However, timing of injury and the presence of additional diseases in patients may influence this result, and the number of patients could be underpowered to detect other differences.
There are a number of neutrophil independent mechanisms that could have influenced our results. First, resolution of lung injury also involves clearance of oedema and restoration of the normal integrity of the alveolocapillary barrier.27 However, we did not find differences between neutropenic and control animals in wet to dry weight ratios or BALF albumin content, a marker of this phenomenon. This could be due to the fact that restoration of barrier function occurs early in the repair process after VILI12 and our observation after 48 hours may be a late time point. Additionally, reabsorption of oedema may increase protein concentration in the alveolar space of the healing lung,28 however, it will have little impact on protein concentration in BALF. Second, our experimental model could have also depleted circulating proinflammatory Gr1+ monocytes that have been implicated in VILI.29 Although it has been reported that monocyte depletion ameliorates rather than deteriorates lung injury,30 selective neutrophil depletion in mice using a specific Ly6G antibody results in a more moderate increase in cytokines,31 suggesting a role for monocytes in immunomodulation. However, neutropenia in patients is commonly related to abnormalities in other cell populations. Upregulation of cytokines during neutropenia has been described also in serum from septic patients,32 and related to the activation of TNF dependent pathways.33 However, previous data showed that blockade of TNFα or MIP-2 has no effect in a model of wound closure using lung epithelial cells,12 and MMP-9 treatment completely reverted the effect of Gr1+ depletion on repair, both in vitro and in vivo without modifying cytokine levels. The close correlation between the experimental and clinical data reinforces the validity of our findings. Moreover, and consistent with our findings, depletion with a Ly6G antibody caused a significant decrease in protease levels in a different model of lung injury.21
Our results may have important clinical consequences. First, the implication of neutrophils in lung repair provides a novel mechanistic explanation for the poor outcome of neutropenic patients when subjected to mechanical ventilation and the failure and risks of anti-inflammatory therapies once injury has been established. Second, it shows a new therapeutic strategy to improve the repair phase. Although treatment with exogenous MMP-9 could cause tissue disruption, other strategies such as growth factors that increase endogenous MMP-9 levels34 could be an alternative in specific groups of patients.
In conclusion, we have shown in a clinically relevant experimental model and in patient samples that neutropenia impairs lung repair. Our results also identify release of MMP-9 from neutrophils as a key mechanism that underlies this phenomenon, and point towards new therapeutic strategies aimed to promote repair after lung injury.
References
Footnotes
JB-P and IL-A contributed equally.
Contributors JB-P, IL-A and GMA designed the study and analysed the data. JB-P, IL-A, LA-R, CH and AG-L performed the experiments. JB-P, IL-A, WMK and GMA discussed the results and wrote the manuscript.
Funding Supported by Instituto de Salud Carlos III (Plan estatal I+D+i, PI16/1614, FEDER funds). IL-A is the recipient of a grant from Fundación para el fomento en Asturias de la investigación científica aplicada y la tecnología (FICYT, GRUPIN14-089, Consejería de Hacienda, Principado de Asturias, Spain). CH is the recipient of a grant from Instituto de Salud Carlos III (Contratos Sara Borrell, CD16/00033). LA-R is the recipient of a grant from Instituto de Salud Carlos III (Contratos Rio Hortega, CM16/00128). GMA is the recipient of a grant from Instituto de Salud Carlos III (INT-15/002). Instituto Universitario de Oncología is supported by Fundación Bancaria Caja de Ahorros de Asturias.
Competing interests None declared.
Ethics approval The Animal Research Ethics Committee of the Universidad de Oviedo evaluated and approved the study. The patient part of the protocol was reviewed and approved by the regional ethics committee (Comité Ético de Investigación Clínica del Principadode Asturias, Spain).
Provenance and peer review Not commissioned; externally peer reviewed.