Article Text

Abstract
Rationale Primary ciliary dyskinesia is a genetically heterogeneous inherited condition characterised by progressive lung disease arising from abnormal cilia function. Approximately half of patients have situs inversus. The estimated prevalence of primary ciliary dyskinesia in the UK South Asian population is 1:2265. Early, accurate diagnosis is key to implementing appropriate management but clinical diagnostic tests can be equivocal.
Objectives To determine the importance of genetic screening for primary ciliary dyskinesia in a UK South Asian population with a typical clinical phenotype, where standard testing is inconclusive.
Methods Next-generation sequencing was used to screen 86 South Asian patients who had a clinical history consistent with primary ciliary dyskinesia. The effect of a CCDC103 p.His154Pro missense variant compared with other dynein arm-associated gene mutations on diagnostic/phenotypic variability was tested. CCDC103 p.His154Pro variant pathogenicity was assessed by oligomerisation assay.
Results Sixteen of 86 (19%) patients carried a homozygous CCDC103 p.His154Pro mutation which was found to disrupt protein oligomerisation. Variable diagnostic test results were obtained including normal nasal nitric oxide levels, normal ciliary beat pattern and frequency and a spectrum of partial and normal dynein arm retention. Fifteen (94%) patients or their sibling(s) had situs inversus suggesting CCDC103 p.His154Pro patients without situs inversus are missed.
Conclusions The CCDC103 p.His154Pro mutation is more prevalent than previously thought in the South Asian community and causes primary ciliary dyskinesia that can be difficult to diagnose using pathology-based clinical tests. Genetic testing is critical when there is a strong clinical phenotype with inconclusive standard diagnostic tests.
- primary ciliary dyskinesia
- respiratory tract
- cilia
- diagnosis
- CCDC103
- mutation
- genetic testing.
Statistics from Altmetric.com
Key messages
What is the key question?
Can gene sequencing improve diagnosis of the inherited respiratory condition primary ciliary dyskinesia (PCD) in patients with unclear clinical diagnostic investigations?
What is the bottom line?
CCDC103 p.His154Pro missense mutations cause up to 20% of PCD cases in South Asian populations but diagnosis can be difficult in this group using standard clinical diagnostic tests because results are often normal; we therefore propose that genetic analysis is an essential part of the diagnostic algorithm to complement standard clinical tests to improve diagnostic accuracy.
Why read on?
Patients with PCD may be missed using current pathology-based diagnostic protocols therefore genetic screening can provide valuable support in obtaining a definitive diagnosis.
Introduction
Primary ciliary dyskinesia (PCD; OMIM: 244400) is an inherited disorder affecting motile cilia. Patients usually present with a history of neonatal respiratory distress and suffer from lifelong symptoms of chronic wet cough and rhinitis. Recurrent chest infections ultimately lead to bronchiectasis and a progressive decline in lung function.1 Approximately half of patients have situs inversus and other situs abnormalities, due to randomisation of situs determination caused by ciliary dysmotility in the embryonic node.2 Fertility can also be affected by defective cilia in the fallopian tubes and non-motile sperm tail flagella.
The estimated prevalence of PCD in the UK is 1:15 000, but as high as 1:2265 in the UK South Asian population.1 3 Early diagnosis is important to maintain lung function, and appropriately treat symptoms to reduce morbidity and mortality.4 Diagnosis can be complex and requires a combination of tests for cilia functional and ultrastructural defects.5 6 PCD is caused by mutations in genes encoding proteins conferring structure and motility to the cilia and governing ciliogenesis. It is genetically heterogeneous with >200 individual mutations in more than 30 genes known to cause PCD. To date, these account for approximately 65% of cases.1 7–11 UK Genetic Testing Network approved tests are offered in two centres (http://www.labs.gosh.nhs.uk/media/764464/ciliopathies_v8.pdf).12
CCDC103 mutations were first reported, in 2012, in patients with dynein arm loss and a typical clinical PCD phenotype.13 CCDC103 is an oligomeric coiled-coil domain protein that is found tightly bound to the ciliary axoneme where it is thought to help facilitate ciliary motility by participating in attachment of the dynein arms to the axoneme. The protein was found to stabilise cytoplasmic microtubules against cold depolymerisation in an in vitro assay.13 A missense variant previously identified in CCDC103 to cause a single amino acid change to the protein, p.His154Pro (rs145457535) was previously described as a hypomorphic mutation since mutant p.His154Pro CCDC103 induced intermediate partially rescued disease phenotypes when expressed in a zebrafish CCDC103-null model, suggesting some protein function was retained.13 In cilia from p.His154Pro-mutated patients some cilia showed a partial dynein arm defect with the outer dynein arms (ODA) at least partially assembled, compared to loss-of-function CCDC103 mutations that cause complete ODA loss. In agreement with this, loss-of-function CCDC103 mutation patients had largely static cilia, while patients carrying a homozygous p.His154Pro mutation exhibit a mixed (static and motile) ciliary beat pattern.13
We conducted genetic screening of 86 patients with PCD of South Asian, mostly Pakistani, origin, detecting that a significant proportion (19%) were homozygous for CCDC103 p.His154Pro variant. Among these, many were at high risk of being undiagnosed without genetic testing, due to normal diagnostic results obtained in PCD investigations. Using electron microscopy and protein biochemistry we have sought to further determine the pathogenic nature of the p.His154Pro mutation.
Methods
Patient selection
Eighty-six patients of South Asian (primarily Pakistani) descent with clinical signs and symptoms of PCD were identified from the UK National PCD Diagnostic and Management Services at The Royal Brompton Hospital, London, University Hospital Southampton, Birmingham Children’s Hospital, Bradford Royal Infirmary and Leicester General Infirmary.1 14
Genetic screening
All participants gave written informed consent to take part in this study. The protocol was approved by the London Bloomsbury Research Ethics Committee (08/H0713/82). High throughput screening used next-generation sequencing, either whole exome sequencing (WES) or targeted gene panel sequencing. Sequencing and variant identification methods are published for WES12 15 or used custom gene panels (Illumina TruSeq Custom Amplicon or Agilent SureSelect Focused Exome (’clinical exome' proprietary product) and SureSelectXT custom panel design systems) with a standardised variant calling pipeline.16 Sanger sequencing was used for variant confirmation and familial segregations. As shown in table 1, of the 16 p.His154Pro cases, this mutation was detected by WES in case numbers 1–6 and number 9, by use of a ‘clinical exome’ commercial panel in case number 8 and in case numbers 7, 10–16 by use of custom targeted gene panels containing the known PCD genes and other candidate PCD disease genes.
Comparator group
The comparator patient group consisted of 16 of the 86 individuals tested. This group was closely age and gender matched to the CCDC103 p.His154Pro group. All had a dynein arm defects on electron microscopy and all were proven negative for the CCDC103 p.His154Pro mutation.
FEV1 measurements
Spirometry was performed according to American Thoracic Society/European Respiratory Society recommendations.17 Forced expiratory volume in 1 second (FEV1, the standard lung airflow measure) z-scores were calculated using the Global Lungs Initiative parameters.18
Diagnostic tests
Screening and diagnostic testing was performed according to the PCD National Service protocols. Investigations included nasal nitric oxide (nNO), nasal brush biopsies analysed by high speed video imaging (HSVM) for ciliary beat frequency and pattern19 and quantitative electron microscopy for ciliary ultrastructure.20 Additional detail on the method is provided in an online supplementary file. When results were inconclusive or inconsistent, patients were offered repeat testing.
Supplementary file 1
Protein biochemistry on recombinant CCDC103 protein
Site-directed mutagenesis (QuikChange kit, Agilent Technologies, UK) was used to generate the p.His154Pro mutation in an N-terminal His10-tagged Homo sapiens CCDC103 cDNA subcloned into pET16b vector that was synthesised using Escherichia coli codon bias.21 Following transformation into E. coli BL21 (DE3), protein expression was induced by addition of 2 mM isopropyl β-D-thiogalactoside (IPTG) for 2 or more hours. Following sonication, His10-tagged proteins were dissolved in 8 M urea, then very slowly refolded by their dilution into 1 L of 20 mM Tris-Cl pH8.0 150 mM NaCl. Proteins were then purified by Ni2+-affinity chromatography as described previously using 20 mM Tris-Cl pH 8.0, 500 mM NaCl, 250 mM imidazole for elution.21 Samples concentrated by ultrafiltration through Amicon Ultra-4 ultrafiltration units were subject to gel filtration in a calibrated Superose 6 10/300 column attached to an ÄktaPurifier-10 chromatography workstation.21 The mutant protein was very hard to make and only a little was able to be refolded, indeed even the precipitated material was clearly different from the wild-type protein being more ‘sticky.’ Hence, a lower concentration of the mutant protein is apparent in figure 1. This experiment was done at one concentration (the highest we were able to achieve): for wild-type protein this was ~80 µg/mL and for the p.His154Pro protein this was ~30 µg/mL.
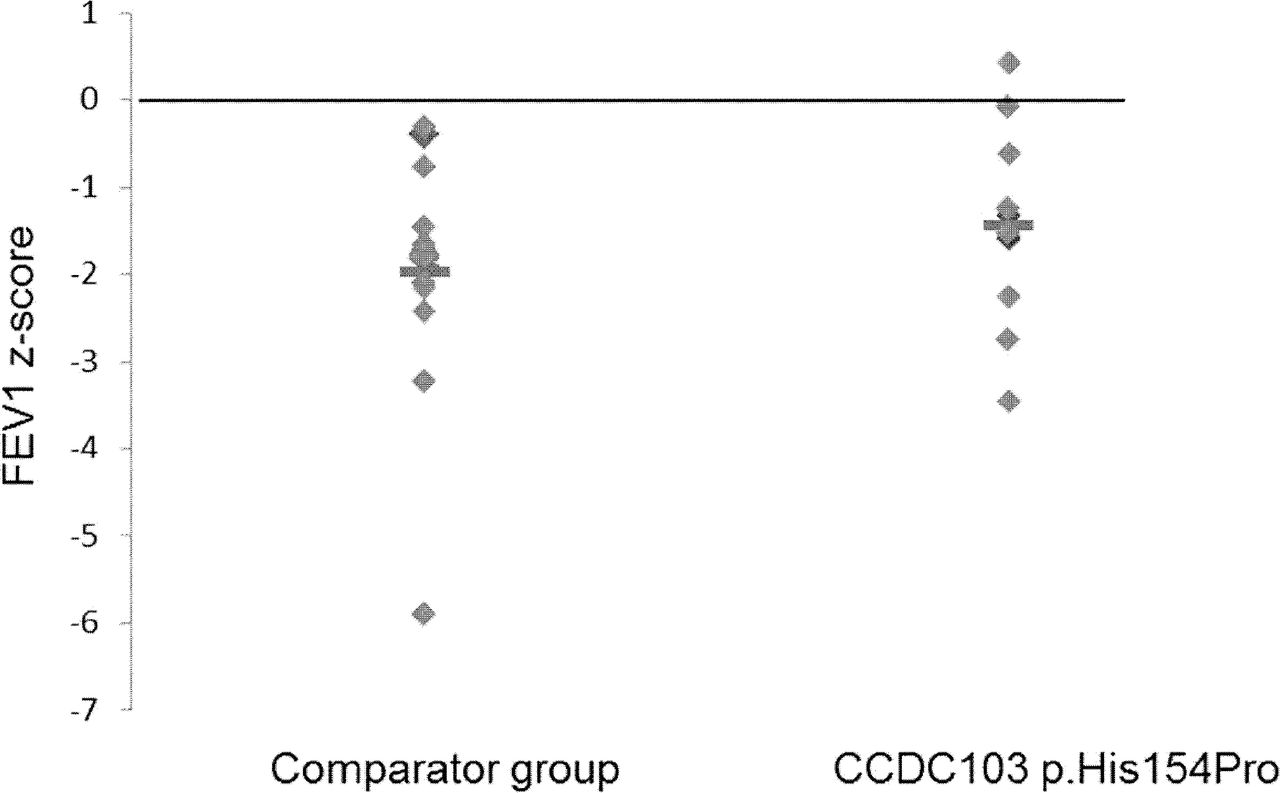
Comparison of predicted FEV1 in CCDC103 p.His154Pro cases versus a comparator group. The CCDC103 p.His154Pro cases are described in table 1 and the comparator group are South Asian origin CCDC103 p.His154Pro-negative cases with a confirmed absent dynein arms defect. Mutations carried by the comparator group are in DNAAF1 (three cases), DNAAF3 (two cases), DNAH5 (two cases), LRRC6 (three cases), ZMYND10 (two cases), while four cases were excluded from CCDC103 p.His154Pro but genetically undefined.
Statistical analysis
The CCDC103 p.His154Pro and comparator patient groups were closely matched for age and gender. Data were not normally distributed and therefore groups were compared using non-parametric statistical tests. p<0.05 was considered statistically significant.
Results
A PCD-causing mutation CCDC103 p.His154Pro is prevalent in UK individuals of South Asian origin with a clinical phenotype of PCD
Next-generation sequencing analysis in 86 patients of South Asian descent with suspected PCD revealed 16 patients (19%) from 11 independent families homozygous for a previously published single base change (NM_001258395.1: c.461A>C) mutation in CCDC103 that predicts the amino acid substitution p.His154Pro (table 1). Consistent with their homozygous segregation pattern, all 16 patients were children of consanguineous parents and had dynein arm defects or normal ultrastructure.
Due to the high frequency of this mutation, we examined whole exome sequence data available from 1542 unaffected parents with similar ethnic backgrounds participating in the Born-in-Bradford study, all of UK South Asian, primarily Pakistani heritage.22 This revealed six heterozygous carriers of the CCDC103 p.His154Pro substitution (E Sheridan, unpublished data). The ExAc database23 of exome sequencing results from 60 706 unrelated control individuals records an allele frequency for p.His154Pro three times as high in 8256 South Asian individuals (0.003) compared with 33,345 North Europeans (0.001). No p.His154Pro homozygote individuals were identified in the entire Born-in-Bradford or ExAc control cohorts.
Among the 16 CCDC103 p.His154Pro homozygote PCD patients identified, normal diagnostic test results were apparent as highlighted in bold in table 1. Although PCD was strongly suspected in all cases, 5 of the 16 patients (case 9 and 13–16 in table 1) did not receive a definitive disease diagnosis until their genotype was confirmed. Remarkably in the case of patient 15, this individual had been deemed to not have PCD and was discharged from respiratory care. She was retested due to her situs inversus and the finding that her brother, who remained under clinical suspicion, had this CCDC103 mutation. Thirteen of the 16 CCDC103 p.His154Pro patients (81%) have situs inversus, including in two families the presence of two siblings with situs inversus, and no families without situs inversus. Situs inversus is reported to affect approximately 50% of patients with PCD with dynein arm defects2 and affected 56% of the PCD comparator group in this study (table 2). Seventy per cent of the comparator group had situs inversus in their family among affected individuals.
Comparison of mean values of clinical tests obtained from CCDC103 p.His154Pro cases versus a comparator group
FEV1 measurements showed that impact on lung function was similar in the group of 16 p.His154Pro patients compared with the PCD comparator group (table 1, figure 1). The comparator group of 16 South Asian individuals with PCD all had dynein arm deficiency, but due to different genetic causes since this group comprised three DNAAF1,24 two DNAAF3,25 two DNAH5,26 three LRRC627 and two ZMYND1028 cases while four were excluded from CCDC103 p.His154Pro but genetically undefined. The mean difference for clinical measures between the CCDC103 p.His154Pro and comparator groups is presented in table 2, showing an equivalent age and gender composition. Individuals from both groups had symptoms typical of PCD as detailed in online supplementary table 1. There was a higher rate of glue ear and positive sputum microbiology and a lower rate of bronchiectasis in the CCDC103 p.His154Pro group compared with controls, but due to the small number of patients and heterogeneous nature of PCD it is difficult to interpret the significance of these findings.
PCD caused by the CCDC103 p.His154Pro mutation can be associated with normal nNO results
We proceeded to retrospectively analyse clinical phenotypes of the 16 affected South Asian patients carrying the homozygous p.His154Pro missense mutation in more detail, as presented in table 1. Of the 16 p.His154Pro patients tested, 7 (43%) had normal nNO levels, above 77 nL/min which is a recommended diagnostic cut-off.29 Of these seven patients, most (patients 5, 7, 11, 12 and 13) were subject to repeat measurements at subsequent clinic appointments. Patient 12 displayed persistent values within the normal range up to 5 years from the first measurement. Mean nNO was significantly higher than the low levels consistently detected in the comparator PCD patient group (table 2).
PCD caused by the CCDC103 p.His154Pro mutation can be associated with areas of normal ciliary beat frequency on high-speed video light microscopy
High-speed video microscopy revealed that among the 16 homozygous p.His154Pro individuals, 9 (56%) had ciliary beat frequencies within the local normal range of 8.5–16.8 Hz (mean 11.6 Hz) (table 1). The other seven p.His154Pro patients (patients 1–6 and 13) had a reduced beat frequency with a mean of 3.9 Hz (range 0–7.7 Hz), which is more in keeping with typical results found for patients with PCD and more specifically found for CCDC103 patients expressing loss-of-function alleles.13 30 The comparator group of patients with PCD displayed a reduced ciliary beat frequency with a mean of 1.3 Hz (table 2), which is similar to the reported reduced ciliary beating seen in published PCD cases associated with other causes of dynein arm loss. In half of the p.His154Pro patients the beat pattern of cilia was completely or largely normal in some strips of epithelium (patients 5–7, 9, 10, 12, 15, 16; table 1) while static, slow or dyskinetic in others. Some p.His154Pro patients (7, 10, 16) demonstrated a full beat pattern and almost immotile cilia together within the same sample (online supplementary videos 1–3). Therefore, the motility in p.His154Pro individuals was significantly different from controls but variable, compared with the fully motile cilia of healthy controls (online supplementary video 4) and the static cilia seen in patients from the comparator group (online supplementary video 5).
Supplementary file 2
Supplementary file 4
Supplementary file 5
Supplementary file 6
PCD caused by the CCDC103 p.His154Pro mutation can be associated with normal ultrastructural appearance by electron microscopy
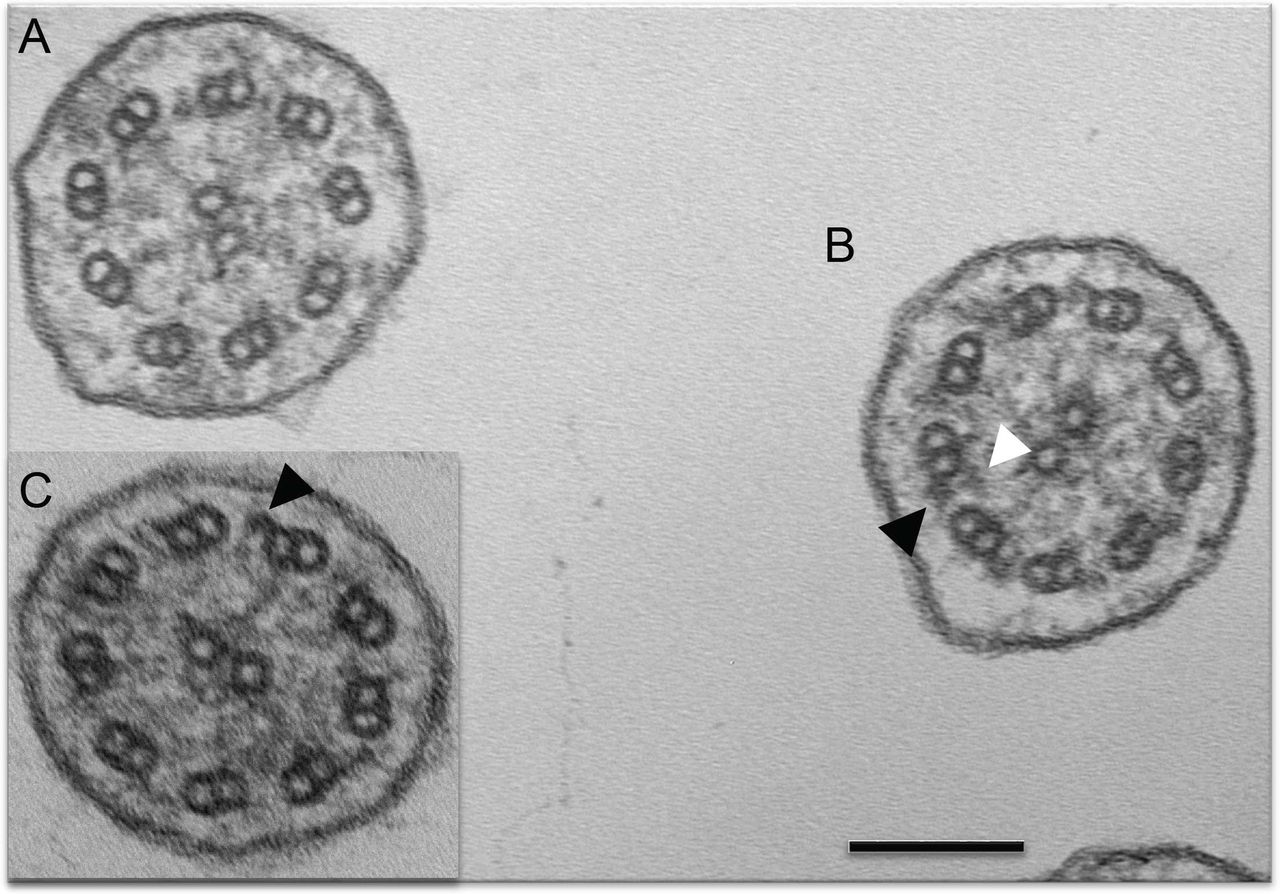
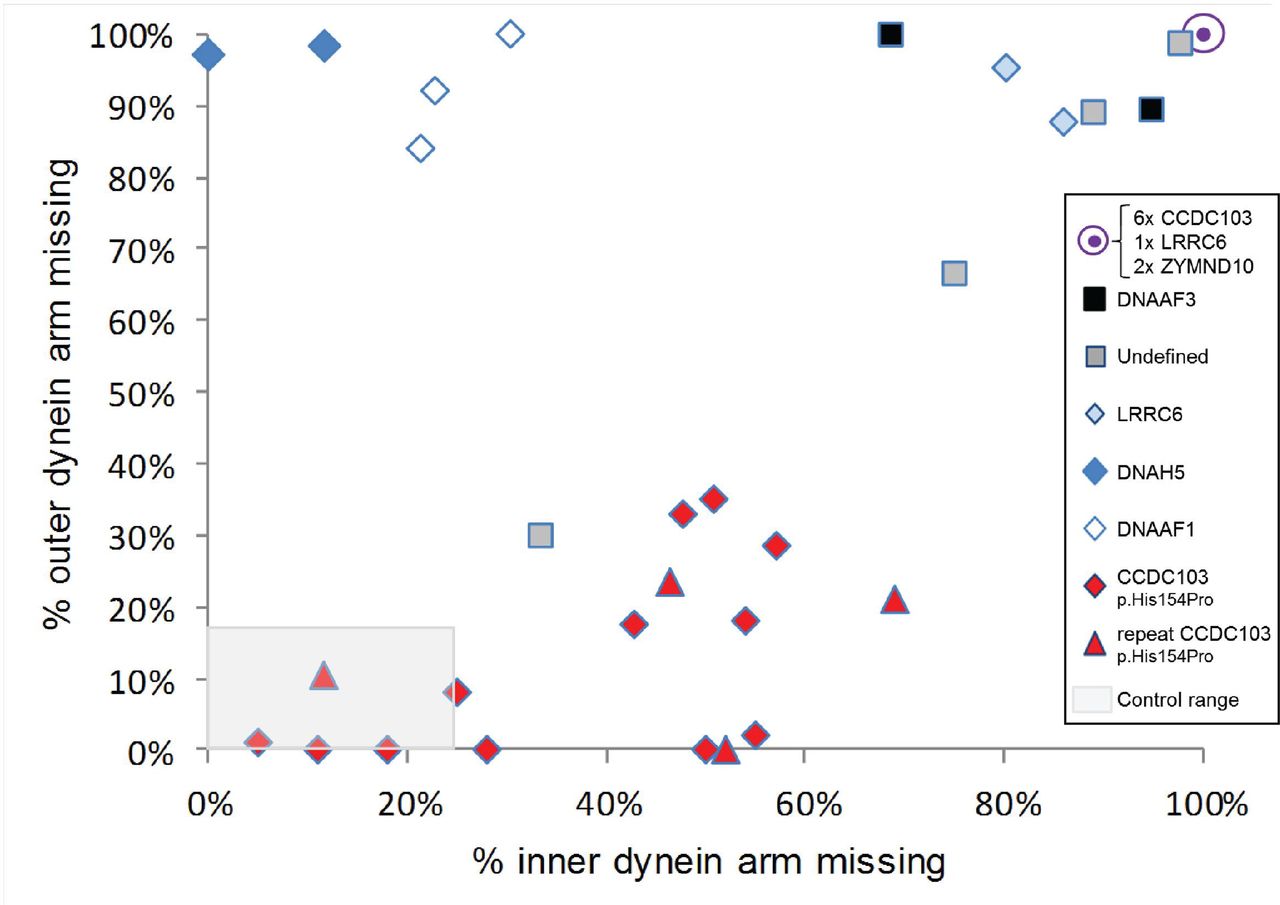
Seven of the 16 CCDC103 p.His154Pro individuals (44%) had a defect of the ciliary inner dynein arms (IDA) and ODA demonstrated by transmission electron microscopy (TEM; cases 1–7 in table 1). The other nine p.His154Pro individuals (cases 8–16) had TEM that either showed an absence of the IDA, or that was considered normal or inconclusive despite extensive interrogation. The spectrum of ultrastructural defects found in p.His154Pro individuals is illustrated with representative examples in figure 2. Overall, the IDAs appeared to be the most affected structure in these cases, with significant retention of ODAs. Quantification of these dynein arm defects is shown in figure 3. Published normal range counts from >200 non-PCD respiratory controls are also shown for comparison (figure 3 grey box). Notably, samples from four CCDC103 p.His154Pro patients were completely within this normal range, a further two were closer to the normal range than the diagnostic range. Two more, although not quantifiable due to small numbers, were also reported as normal (not shown). When a partial defect of the ODA was seen in p.His154Pro patients, an assessment of proximity to the epithelial cell surface—as judged by the presence of neighbouring microvilli—showed that ODA loss typically occurred at the distal end of the cilia, towards the tips.

Transmission electron microscopy of CCDC103 p.His154Pro patients. Representative cilia cross sections show within the same micrograph (A) absent outer and partially absent inner dynein arms and (B) presence of outer dynein arms only and presence of both inner and outer dynein arms. (C) Inset shows presence of outer but not inner dynein arms. Black arrows indicate example of outer dynein arms. White arrows indicate inner dynein arms. Scale bar, 100 nm.
Clinical history of CCDC103 p.His154Pro cases summarising diagnostic investigations

Quantitative transmission electron microscopy survey of inner and outer dynein arm loss in CCDC103 p.His154Pro patients versus a comparator group. By surveying >100 cross sections in each patient sample we performed quantitative electron microscopy to determine the percentage of arm defects in cilia from individuals homozygous for the hypomorphic p.His154Pro CCDC103 mutation. Note that quantitative plots have only been included within this graph if more than 100 cilia were counted and that all data have been collected at a single centre to ensure uniform results; data collected for one sample from a separate centre were therefore excluded. Red diamonds and triangles indicate results from 16 CCDC103 p.His154Pro homozygote PCD patients, where triangles indicate the result of four repeat nasal brushings performed on patients marked by a diamond. The comparator group of 16 individuals with PCD, indicated by other symbols as shown, consists of three cases with DNAAF1 mutations (open diamonds), two DNAAF3 (black squares), two DNAH5 (dark blue diamonds), three LRRC6 (two as light blue diamonds, one contained within the filled purple circle), two ZMYND10 (contained within the filled purple circle) and four cases in whom no mutations in CCDC103 or other known genes could be identified (grey squares). Six CCDC103 p.His154Pro samples (27%) of p.His154Pro samples showed complete lack of both outer and inner dynein arms comparable to other gene mutations in the graph (purple circle). The grey shaded area represents normal range counts from >200 non-PCD respiratory controls. Four CCDC103 p.His154Pro patients had counts within this normal range (one is a repeat sample (triangle) which showed similar data). Individuals with CCDC103 p.His154Pro mutations have a trend towards a distinctive pattern of partial loss of dynein arms that diverges from total dynein arm loss in the comparator group.
A summary shown in table 2 highlights that the majority of CCDC103 p.His154Pro cases clearly differed from the non-CCDC103 comparator group, which have a near complete absence of both dynein arms. This is highly significant in terms of ODA loss (p<0.05), with only 19% ODA absence in the CCDC103 p.His154Pro cilia compared with 89% in the comparator group which carry mutations in other dynein arm loss-associated PCD genes. IDAs were also clearly more retained in CCDC103 p.His154Pro cilia, but this difference did not reach statistical significance. The overall relative lack of disturbance to dynein arm structures means that CCDC103 p.His154Pro TEM overlaps with that of non-PCD controls.
Biochemical analysis of the CCDC103 p.His154Pro mutation reveals an abrogation of its oligomerisation capacity
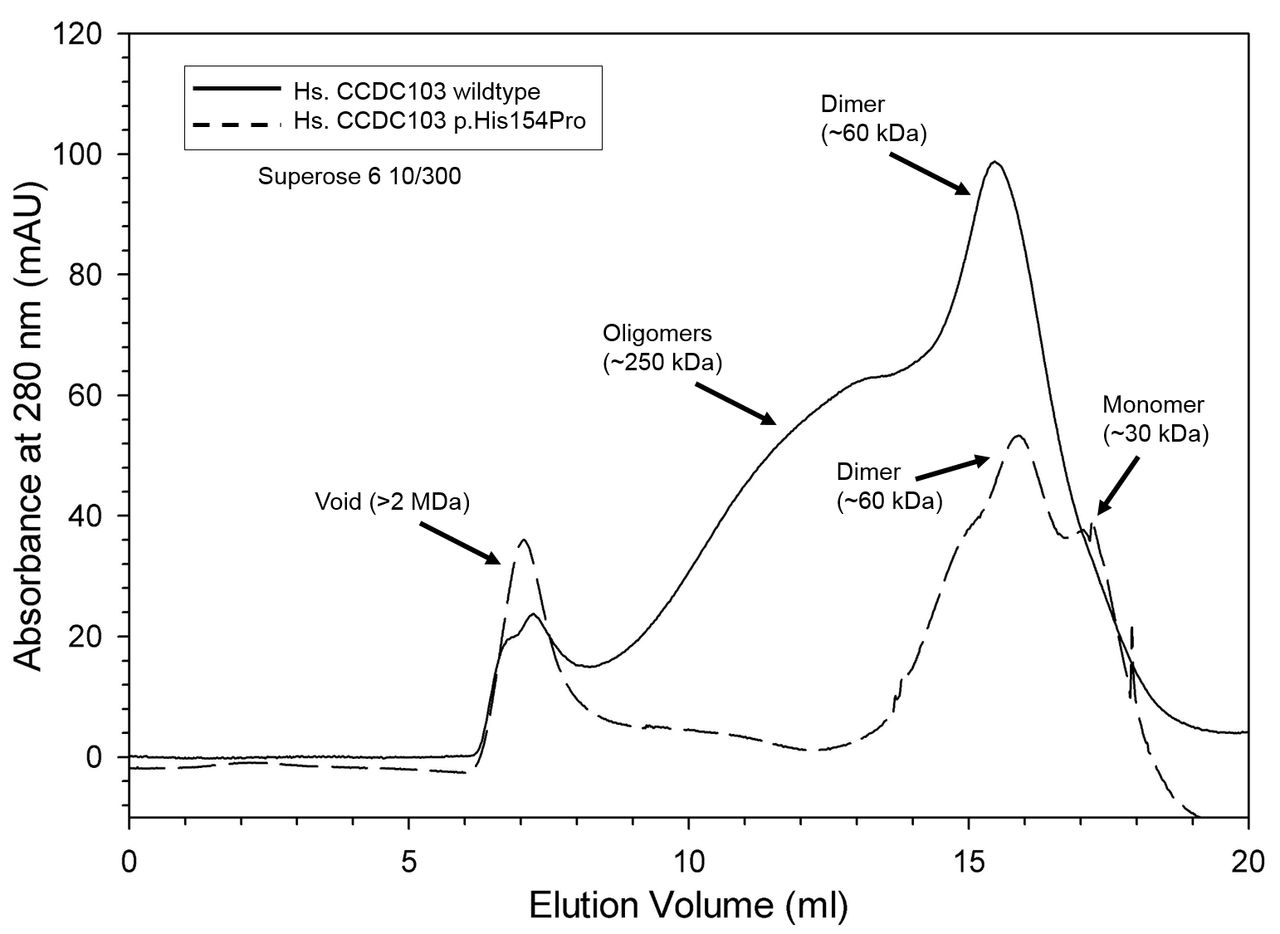
We conducted gel filtration of purified CCDC103 p.His154Pro protein and compared this to the normal protein using previously established methods21 to assess the functional viability of the mutant form of the protein. The p.His154Pro variant appears to be a highly disruptive mutation since the ability of CCDC103 to oligomerise is significantly disrupted by the mutation, as shown in the chromatogram (figure 4). The lack of oligomers that the wild-type protein forms (~250 kDa) is however accompanied by a large increase in the void volume of the mutant protein. We speculate that this material, which is >2 MDa, is aggregated protein, rather than organised oligomers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CCDC103 p.His154Pro oligomerisation capacity chromatograms of wild-type (solid trace) and His154Pro (dashed trace) CCDC103 native proteins separated in a calibrated Superose 6 10/300 gel filtration column. The data are plotted as absorbance at 280 nm (in mAU) against elution volume (mL). Both proteins show strong dimer peaks at ~60 kDa. However, only the wild-type form generates a series of higher order oligomers with an approximate mass of ~250 kDa. Aggregated material (>2 MDa) eluted in the void volume.
Discussion
We report from genetic screening of a PCD cohort ascertained through UK National PCD Services that the CCDC103 c.461A>C; p.His154Pro mutation accounts for disease in approximately one-fifth of affected individuals in the highly consanguineous South Asian PCD community, associated with loss of both the ODA and IDA of cilia. Therefore, a significant risk of disease arises from the presence of this important frequent mutation which is typically carried in homozygous state, one copy inherited from each parent. Direct screening for this specific p.His154Pro mutation in South Asian PCD cases could be an economical diagnostic approach.
Despite the relatively high prevalence of CCDC103 p.His154Pro, this mutation is not the sole cause of the increased incidence of PCD reported in the British South Asian community.3 Our screen also revealed patients in the group of 86 screened that carry causal mutations in a number of other genes including CCDC40, DNAAF1, DNAAF3, HEATR2, LRRC6, ZMYND10 and RSPH4A. The CCDC103 p.His154Pro defect is not exclusive to the UK Asian community and has also been detected in a patient from an Irish travelling family31 as well as in two North European origin families with PCD from our studies (unpublished data), all of whom have dynein arm defects.
This study highlights extensive variability in the diagnostic results for patients carrying biallelic CCDC103 p.His154Pro mutations. Normal standard diagnostic test results have led to CCDC103 p.His154Pro patients being discharged from the PCD clinic, and in some cases the diagnosis of PCD in a CCDC103 p.His154Pro patient was only finally confirmed following the genetic test result. Fifteen (94%) patients had situs inversus, suggesting CCDC103 p.His154Pro patients without situs inversus may be missed either due to lack of referral or due to normal diagnostic tests.
The homozygous p.His154Pro-positive patients represent an expanded phenotype for PCD, since without genetic results many in this group may not have been considered to meet the current UK clinical diagnostic criteria for PCD. This would lead to uncertainty for the patient and their parents and may result in unnecessary further investigation into the cause of their symptoms. One South Asian patient in the comparator group had a TEM phenotype similar to that of the CCDC103 p.His154Pro group. This individual also had an nNO level of >77 nL/min, but a screen for the CCDC103 mutation was negative (figure 3, lower left grey square). This case, along with other patients in the UK national diagnostic clinics, suggests that there will be other mutations which cause PCD with normal diagnostic results.
It is difficult to explain the variability in the p.His154Pro patients nNO (table 1). This variability is temporal within a patient and seen even between siblings. We speculate that this may be a broader reflection of ciliary function in the sinuses such that NO levels may only be low in CCDC103 p.His154Pro patients when cilia are static (maybe during an infection or other external insult) and may normalise with improved ciliary function. The possible reasons why levels might be low in PCD were recently summarised.32
Interestingly, preservation of some cilia structure and motility in CCDC103 p.His154Pro patients is not apparently accompanied by significantly preserved lung function, since the FEV1 range in p.His154Pro patients is equivalent to the comparator group and respiratory capacity is overall equally reduced in these individuals. Another missense mutation causing PCD, ZMYND10 p.Val16Gly, was similarly found to cause a mixed cilia beat defect with a significant degree of retained cilia motility but this is also not yet associated with milder disease course.28 In future such studies, larger numbers of CCDC103 p.His154Pro patients and controls should be analysed to confirm this observation and genotype— phenotype relationships could be further investigated with more sensitive tests for staging lung disease such as Lung Clearance Index, CT scan or radiolabelling methods screening mucociliary clearance rates.33 34
Patients with the CCDC103 p.His154Pro mutation represent an interesting cohort for targeted pharmacogenetic therapies due to their prevalence and since the dynein components required for normal ciliary beating appear to still be present in the patient’s cilia, although at variable levels.
The finding of nNO levels within the normal range in CCDC103 p.His154Pro patients highlights the importance of considering the full clinical history in conjunction with nNO testing in patients with suspected PCD. Clinicians should proceed to further testing in cases with a high index of clinical suspicion, using the NO test as part of a multidisciplinary diagnostic protocol rather than a stand-alone screening test. This is not the first report of normal nNO results in patients with PCD. Some patients with RSPH1 mutations are also reported to have levels of NO close to normal and we expect further such cases to be reported as the complex genetic landscape of PCD diagnosis is unravelled further.35
The apparently normal/mixed cilia beat pattern found in half of p.His154Pro cases was notably high, but this finding is supported by previous work indicating that this represents a hypomorphic allele in cilia function tests.13 Awareness of these cases is critical when assessing diagnostic samples in the laboratory, if the observer inadvertently tends to select beating strips for analysis over those that are static when scanning the sample at 5–20 times magnification before full analysis with high-speed video, thereby inadvertently missing this defect when a significant portion of the sample has a normal coordinated beat. This places an emphasis on diagnostic centres to increase awareness and expertise of the operators when assessing nasal biopsies by high-speed video microscopy. HSVM does not quantify the power of the ciliary beating, but in cases with a normal stroke and frequency there could be weakness in the strength of the beating that might not be detected by any of the current diagnostic tests.
In this study, TEM diagnosis often revealed a pattern of intermittent IDA and ODA loss in CCDC103 p.His154Pro patients which was distinct from cases of dynein arm absence due to mutations in other dynein loss-associated PCD genes, for example, LRRC6, DNAAF1 and ZYMND10. The distinction was most significant for ODA retention with CCDC103 p.His154Pro cases, with comparatively high levels of preserved ODAs seen in patient’s cilia while the IDAs were more often missing. Despite analysis of >100 cilia cross sections, usually sufficient for diagnosis, the loss of dynein was not always detected by electron microscopy. However, it has long been accepted that normal ultrastructure by electron microscopy cannot exclude PCD.36 37 Our study appears to confirm in CCDC103 p.His154Pro patients the previous evidence that CCDC103 mutations confer a loss of distal ODAs containing DNAH9, not ODAs in the proximal half of the cilia closer to the epithelial surface.13
This study contributes strong evidence that CCDC103 p.His154Pro is pathogenic rather than a benign polymorphism. It is present at very low frequency in the non-PCD population and no CCDC103 p.His154Pro homozygote individuals were detected in large-scale screening of 9798 South Asian and 33,345 North European controls. Our segregation analysis performed where parental samples were available in the affected families studied here showed an inheritance pattern fully consistent with recessive disease (table 1). The CCDC103 protein remains poorly characterised but previous studies show that it usually forms dimers and higher order oligomers. It is thought to help in generating a high-affinity site on the doublets for outer arm assembly, either through direct interactions or indirectly, by modifying the underlying microtubule lattice.21 The oligomerisation capacity of CCDC103 is a property of the central region of the protein (RSPK and SMK, unpublished observations) which contains a highly conserved RPAP3_C domain that spans residues 96–189 of the protein and is predicted to function in protein–protein interactions.21 The His154 amino acid is located within an alpha helix of this RPAP3_C functional domain and substitution of the cyclic side chain of proline at this position would enforce a conformation predicted incompatible with alpha helical secondary structure. Thus, altering His154 is expected to disrupt the protein’s secondary structure and consequently its function. Our current results reveal that the His154Pro mutant form retains the ability to dimerise but shows little oligomer formation, suggesting that this property is disrupted by the mutation and that CCDC103 may have two distinct self-interaction domains. We hypothesise that gene mutations causing instability or depletion of CCDC103 protein may make the attachment of the dynein arms more susceptible to physical, infective or inflammatory insult. We cannot determine if the presence of normal cilia in our patients is temporal or spatial; however, as results appear to vary from one biopsy to the next, for example, in TEM data for cases 11, 12, 15 and 16 in table 1, therefore cell culture and repeating investigations may be useful in these cases.
It can often be difficult to make or exclude a diagnosis of PCD due to poor sensitivity of electron microscopy and genetic testing and the poor specificity of nNO measurement.5 Currently, diagnosis using multiple tests is recommended. These tests are often complex and require specialist equipment and skills to interpret. Consequently, several patients continue to have an indeterminate diagnosis. Awareness of variants such as CCDC103 p.His154Pro may allow targeted gene screening in patients with an indeterminate diagnosis.
In conclusion, the CCDC103 p.His154Pro variant is prevalent in the UK South Asian community and likely to be found in South Asian patients worldwide. This patient group should therefore undergo genetic testing for c.461A>C, especially if (partial) dynein arm absence is suspected. These patients frequently present a diagnostic dilemma, due to inconclusive results of multiple clinical diagnostic tests. This study expands the diagnostic phenotype which we consider to be PCD, since in some cases pathology-based tests can be equivocal as the cilia can beat at least partially in a coordinated manner, at the correct speed and may appear structurally normal. PCD is widely understood to be an underdiagnosed condition and this appears to be the case for CCDC103 p.His154Pro patients, who demonstrate a high level of situs inversus probably indicative of a lack of recognition making their diagnosis liable to be missed.38 We anticipate that studies such as this, in combination with easier access to high throughput and economically achievable genetic screening, should greatly increase disease recognition and understanding. We have highlighted the importance of multidisciplinary testing, repeat testing and genotyping in patients with a highly suggestive history for PCD.
Supplementary file 3
Acknowledgments
We are very grateful to the families with PCD who have participated in this study and to the UK PCD Family Support Group for their support. We thank Louise Ocaka, Chela James (UCL GOSgene) and Emily Frost (Royal Brompton Hospital) for experimental support and data analysis.
References
Footnotes
Contributors AS, HMM, EAR and EM were responsible for study concept and design. MPP, JH, JS, MRF, WL, HW, CB, LJ, CMW, TC and HMM performed the genetic analyses. RSPK and SMK performed the biochemical analysis. AS, MD, AVR, AR and PG performed electron microscopy. AS, RAH, MD, AVR, CLJ and SO performed cilia cell culture and cilia functional testing. EM, EAR, SC, WW, BA, MRL, BR, RW, AB, ES, JSL, PK, COC and CH provided clinical information. AS, SMK, CH and HMM wrote the manuscript and all authors contributed to the final version. HMM is the guarantor responsible for the overall content.
Funding The research is supported by the BEAT-PCD: Better Evidence to Advance Therapeutic options for PCD network (COST Action 1407). AB was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. Work by AS is independent research funded by a postdoctoral research fellowship from the National Institute of Health Research and Health Education England. RSPK and SMK are supported by NIH grant GM051293. MS is supported by a Radboudumc Hypatia Tenure Track fellowship, a Radboud University Excellence fellowship, an ERC starting grant (TREATCilia, grant no.716344) and received funding from the German Research Foundation (DFG), collaborative Research Center (CRC) 1140 KIDGEM. This research and the Centre for Translational Omics (GOSgene) is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. HMM was supported by grants from Action Medical Research (GN2101), Newlife Foundation (10-11/15) and the Great Ormond Street Hospital Children’s Charity. Work in Southampton is supported by the NIHR Respiratory Biomedical Research Unit and NIHR Wellcome Trust Clinical Research Facility.
Competing interests None declared.
Ethics approval London Bloomsbury Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.