Article Text

Abstract
Background Obstructive sleep apnoea (OSA) is an important cause of secondary hypertension. Nocturnal hypertension is particularly prevalent in OSA and is a strong predictor of cardiovascular mortality. Studies in patients with essential hypertension have suggested that nocturnal administration of antihypertensives improves nocturnal blood pressure (BP) without elevating daytime BP. We evaluated the efficacy of this technique in patients with OSA with stage I/II hypertension, both before and after the addition of CPAP.
Methods In this double-blind randomised placebo-controlled crossover trial, patients with moderate-to-severe OSA and hypertension received 6 weeks each of evening or morning perindopril with opposing time-matched placebo. CPAP therapy was subsequently added for 8 weeks in addition to either morning or evening perindopril. The primary outcome was sleep systolic BP (SBP) using 24-hour BP monitoring, analysed using linear mixed models.
Results Between March 2011 and January 2015, 85 patients were randomised, 79 completed both dosing times, 78 completed the CPAP phase. Sleep SBP reduced significantly from baseline with both evening (−6.9 mm Hg) and morning (−8.0 mm Hg) dosing, but there was no difference between dosing times (difference: 1.1 mm Hg, 95% CI −0.3 to 2.5). However, wake SBP reduced more with morning (−9.8 mm Hg) than evening (−8.0 mm Hg) dosing (difference: 1.8 mm Hg, 95% CI 1.1 to 2.5). Addition of CPAP to either evening or morning dosing further reduced sleep SBP, but by a similar amount (evening: −3.2 mm Hg, 95% CI −5.1 to −1.3; morning: −3.3 mm Hg, 95% CI −5.2 to 1.5).
Conclusions Our findings support combining OSA treatment with morning administration of antihypertensives. Unlike in essential hypertension, our results do not support evening administration of antihypertensives, at least with perindopril. Further research is required before this strategy can be widely adopted into hypertension guidelines and clinical practice.
Trial registration number ACTRN12611000216910, Results.
- Sleep apnoea
Statistics from Altmetric.com
Key messages
What is the key question?
In patients with essential hypertension, randomised trials have demonstrated that chronotherapy involving ingestion of a once daily dose of antihypertensive medication in the evening instead of the usual morning, can achieve a twofold to threefold greater reduction in systolic nocturnal blood pressure without raising daytime blood pressure and so we sought to determine whether this simple and novel strategy could achieve the same dramatic results in patients with obstructive sleep apnoea (OSA) where treatment of hypertension is difficult.
What is the bottom line?
Our randomised placebo-controlled trial in patients with OSA did not find a superior effect on blood pressure control from evening administration of perindopril.
Why read on?
This is the most robust chronotherapy trial thus far performed in any population and our opposing results highlight the complexities and differences of treating hypertension in patients with OSA.
Introduction
Hypertension increases the risk of cardiovascular mortality and early treatment with pharmacotherapy reduces this risk. Obstructive sleep apnoea (OSA) is an important secondary cause of hypertension,1 ,2 with large epidemiological studies showing a strong dose-dependent relationship with both prevalent and incident hypertension.3 ,4 Intermittent hypoxia is thought to be one of the main pathophysiological mechanisms leading to the development of hypertension in sleep apnoea.5 ,6 Meta-analyses indicate that CPAP, the gold standard treatment for OSA, results in only a modest 1–2 mm Hg reduction in blood pressure (BP),7 ,8 in part due to poor long-term adherence with this therapy.9 On the other hand, pharmacotherapy with antihypertensive medication results in much larger BP reductions in patients with sleep apnoea,10 ,11 but not as pronounced as in hypertensive patients without OSA.12 Despite this, in patients with treatment-resistant hypertension, where the use of multiple antihypertensives has failed, the prevalence of unrecognised sleep apnoea is very high, with reports of up to 83%.13 Additionally, patients with both OSA and resistant hypertension appear to respond more avidly to CPAP, with reductions in systolic BP (SBP) of 7–8 mm Hg.14 ,15 These data suggest that stand-alone pharmacotherapy for hypertension in OSA may not be entirely adequate and that CPAP therapy should be added to the treatment regimen where tolerated.
Numerous prospective studies have found that both nocturnal hypertension16–18 and, to a lesser extent, non-dipping19 predict increased cardiovascular mortality. This has fuelled a recent interest in the practice of chronotherapy for hypertension—nocturnal ingestion of antihypertensives to specifically target nocturnal BP. Despite some design limitations, randomised chronotherapy trials in essential hypertension,20 chronic kidney disease,21 diabetes22 and resistant hypertension23 have demonstrated a twofold to threefold greater reduction in systolic nocturnal BP (up to 14.2 mm Hg drops) with evening ingestion of antihypertensives while maintaining the same reductions in daytime BP. These chronotherapy studies have also suggested subsequent reductions in cardiovascular morbidity and mortality.20–23 Patients with OSA may stand to benefit significantly from this simple strategy, particularly given the known high rates of nocturnal hypertension24 ,25 and non-dipping.26–28 Furthermore, the potential effect sizes would be much greater than that achieved with CPAP.
Surprisingly, chronotherapy has never been robustly tested in hypertensive patients with OSA, although a single non-randomised clinical trial found a markedly greater reduction in sleep BP (by 4.4 mm Hg) and increased rates of nocturnal dipping with evening ingestion of antihypertensives.29 Therefore, we conducted a double-blinded randomised placebo-controlled crossover trial in patients with untreated OSA and hypertension. We hypothesised that evening ingestion of the antihypertensive perindopril would result in a larger reduction in sleep SBP than morning ingestion. We further explored whether the addition of CPAP therapy would result in a further reduction in sleep SBP.
Methods
Study design
We compared the effects of evening versus morning administration of once-daily 10 mg perindopril arginine (Coversyl, Servier) with placebo at the opposite time point, on ambulatory BP in patients with moderate-to-severe OSA and stage I–II hypertension. The study was a randomised, double-blind, placebo-controlled crossover design (phases I and II) followed by open-label CPAP (phase III) (figure 1). The study protocol was approved by the Northern Sydney Human Research Ethics Committee (AU RED Ref HREC/10/HARBR/51). All patients signed a written informed consent.

Study design and flow chart of patients in the study. AF, atrial fibrillation; AM, morning dosing; ARB, angiotensin receptor blocker; BP, blood pressure; CSA, central sleep apnoea; eGFR, estimated glomerular filtration rate; anti-HTN, antihypertensive; ODI, oxygen desaturation index; PM, evening dosing; PSG, polysomnogram.
Patients
Patients were recruited from tertiary referral clinics (The Woolcock Clinic, Royal Prince Alfred Hospital, Liverpool Hospital and Royal North Shore Hospital), and private specialist clinics as well as directly from the community via radio and local newspaper advertising, all in Sydney, Australia.
Male and female patients were eligible if they were aged 18–65 years, had moderate-to-severe untreated hypoxaemic OSA (apnoea-hypopnoea index (AHI) and oxygen desaturation index 3% ≥15/hour), stage I or II hypertension based on office BP (SBP 140–179 mm Hg and/or diastolic BP (DBP) 90–109 mm Hg) and elevated BP awake (SBP ≥135 mm Hg and/or DBP ≥85 mm Hg) or asleep (SBP ≥120 mm Hg and/or DBP ≥70 mm Hg) based on 24-hour ambulatory BP monitoring (24-hour ABPM) and on fewer than three antihypertensive medications that did not include an ACE inhibitors or angiotensin receptor blocker (ARB) at randomisation. Patient exclusion criteria were: severe hypertension (office SBP ≥180 mm Hg and/or DBP ≥110 mm Hg), severe OSA (minimum oxygen saturation ≤65% or respiratory disturbance index >80) with excessive sleepiness and at high risk of driving-related accidents, chronic kidney disease (estimated glomerular filtration rate <60) and uncontrolled medical problems. Additional exclusion criteria are outlined in the online supplement.
supplement data
All patients underwent gold standard overnight in-laboratory polysomnography to confirm OSA status and severity. Potential patients who were already on an ACE inhibitor or ARB were withdrawn from these antihypertensive medications (washed out) for 3 weeks prior to baseline assessments. At the baseline eligibility visit, patients had an office BP assessment and then underwent 24-hour ABPM (Spacelabs 90217-1Q ambulatory BP monitor, Spacelabs Medical, Snoqualmie, Washington, USA). Sleep and wake periods were assessed using wrist actigraphy (Actiwatch 2, Phillips Healthcare, Murrysville, Pennsylvania, USA).30 Patients with stage I–II hypertension and ambulatory awake BP ≥135/85 mm Hg and/or sleep BP ≥120/70 mm Hg proceeded to the perindopril run-in phase to ensure medication tolerability. The run-in duration was dependent on previous ACE inhibitor/ARB exposure as outlined in the online supplement. Perindopril at 10 mg is the highest dose in its formulation.
Procedures
After the run-in phase, patients who remained tolerant to the study drug were randomised to either morning dosing of perindopril 10 mg with evening placebo or evening perindopril 10 mg with morning placebo for 6 weeks (phase I). This was followed by crossing over to the alternate regimen for the next 6 weeks (phase II). Patients were instructed to take their trial medications at approximately 09:00 and 21:00. Patients who were taking other classes of antihypertensives (n=42) were asked to continue taking those medications at the same time of day as they had before entering the trial. There was no washout period between the treatment arms because steady state for this drug is achieved within 5–6 days, which is well before the end point assessments. In the final 8 weeks (open-label phase III), patients were treated with CPAP therapy for their OSA in addition to continuing the active dose of perindopril as allocated in phase II of the study.
The settings for CPAP pressure were determined during a single overnight in-laboratory pressure titration sleep study at least 3 weeks prior to the end of treatment phase II. This study involved an initial mask fitting and CPAP education session followed by an overnight study. CPAP pressure was increased manually to a level that abolished all apnoeas and hypopnoeas. Patients were reviewed 1–2 weeks after commencement of CPAP for optimisation of therapy as outlined in the online supplement.
At baseline and at the end of each treatment phase, patients underwent 24-hour ABPM with concurrent actigraphy in the community while completing sleep diaries. We estimated sleep and wake times for each individual patient from actigraphy and sleep diary data (see online supplement). Daytime naps were labelled as ‘sleep’ and wakeful periods at night were labelled as ‘wake’ to ensure that corresponding BP readings were accurately identified, regardless of time of day. Patients who had <80% successful readings on 24-hour ABPM were asked to repeat the testing. BP monitors were validated on application and removal of the device. Medication compliance was assessed using pill counts from returned bottles at the end of each treatment arm.
Outcomes
The primary end point was the difference in sleep SBP determined by 24-hour ABPM between morning and evening ingestion of perindopril. Secondary outcomes included sleep DBP, wake and 24-hour SBP and DBP, BP dipping ratios and office BP. Changes in these outcomes with CPAP were also determined.
Adverse events and safety data were collected throughout the trial. Patients were asked to contact the investigators in the event of any adverse events and were also questioned directly about any potential adverse events at each treatment visit.
Statistical analysis
Based on data from a previous chronotherapy trial,31 we conservatively estimated the within-subject SD of ambulatory SBP to be 9 mm Hg. We calculated that 82 patients would provide 80% power to detect a 4 mm Hg SBP difference between evening and morning dosing.
Analysis was by intention to treat. Linear mixed models using all discrete BP measures were used to examine changes in BP outcomes with different dosing times including order effects, both before and after CPAP. The primary analysis was performed with the active dose time as a fixed effect and the patient as a random effect in all models. For the CPAP open-label analysis, the timing of the active dose (morning or evening), the treatment arm (before or after CPAP) and the interaction of these two binary variables were included as fixed effects.
Comparison of office BP measurements between baseline and the end of morning and evening dosing were determined using paired samples t-tests. CPAP effects were determined using independent samples t-test.
Non-dipping status was calculated as (sleep SBP mean/wake SBP mean)×100 ≤10%. Comparison of non-dipping rates between conditions was performed within or between subjects using McNemar's test or the χ2 test for independent proportions, respectively.
For the exploratory subgroup analysis of non-dippers, the same statistical analyses as above were used with the exception that Fischer's exact test was performed to compare non-dipping rates between subjects due to the small sample size. All statistical analyses were performed using IBM SPSS V.22.0 for Windows system (SPSS, Chicago, Illinois, USA).
This clinical trial is registered with the Australian and New Zealand Clinical Trials Registry at http://www.anzctr.org.au (ACTRN12611000216910).
Randomisation and masking
The allocation of treatment sequence was performed by an independent company (Trident Clinical Research, Melbourne, Australia) according to a randomised block design. It was computer generated by a SAS program (version 8.2) using the RANUNI function and a random seed. These were stored in sequentially numbered sealed opaque envelopes. Patients were sequentially enrolled and assigned the next randomisation number. The placebo and active medication capsules were identical. Patients and all staff involved in outcomes assessments remained blinded to the treatment allocation until after collecting all outcome data at the end of the second treatment phase, immediately prior to the open-label phase III. This final phase with CPAP did not involve placebo ingestion making it necessary for the second treatment allocation to be revealed to patients and staff involved in outcomes collection. Success of masking was not specifically assessed.
Prior to statistical analysis, all 24-hour ABPM data that were deemed artefactual was identified and removed based on Staessen's criteria, which included the following readings: SBP >240 or <50 mm Hg, DBP >140 or <40 mm Hg, HR >150 or <40 bpm and pulse pressure >10% of SBP.32 All statistical analyses of main outcomes were performed blinded to treatment allocation.
Results
Between March 2011 and January 2015, a total of 155 patients were screened and 85 underwent randomisation. A flow diagram detailing the exclusions and the reasons is shown in figure 1. A total of 79 patients completed both medication ingestion times, and 78 patients completed the entire trial. The baseline characteristics of all randomised patients are found in table 1. There was no change in weight in patients from baseline to either morning or evening dosing (see online supplement for further details). All 24-hour BP readings were available for all patients who completed the trial.
Baseline characteristics of all randomised patients*
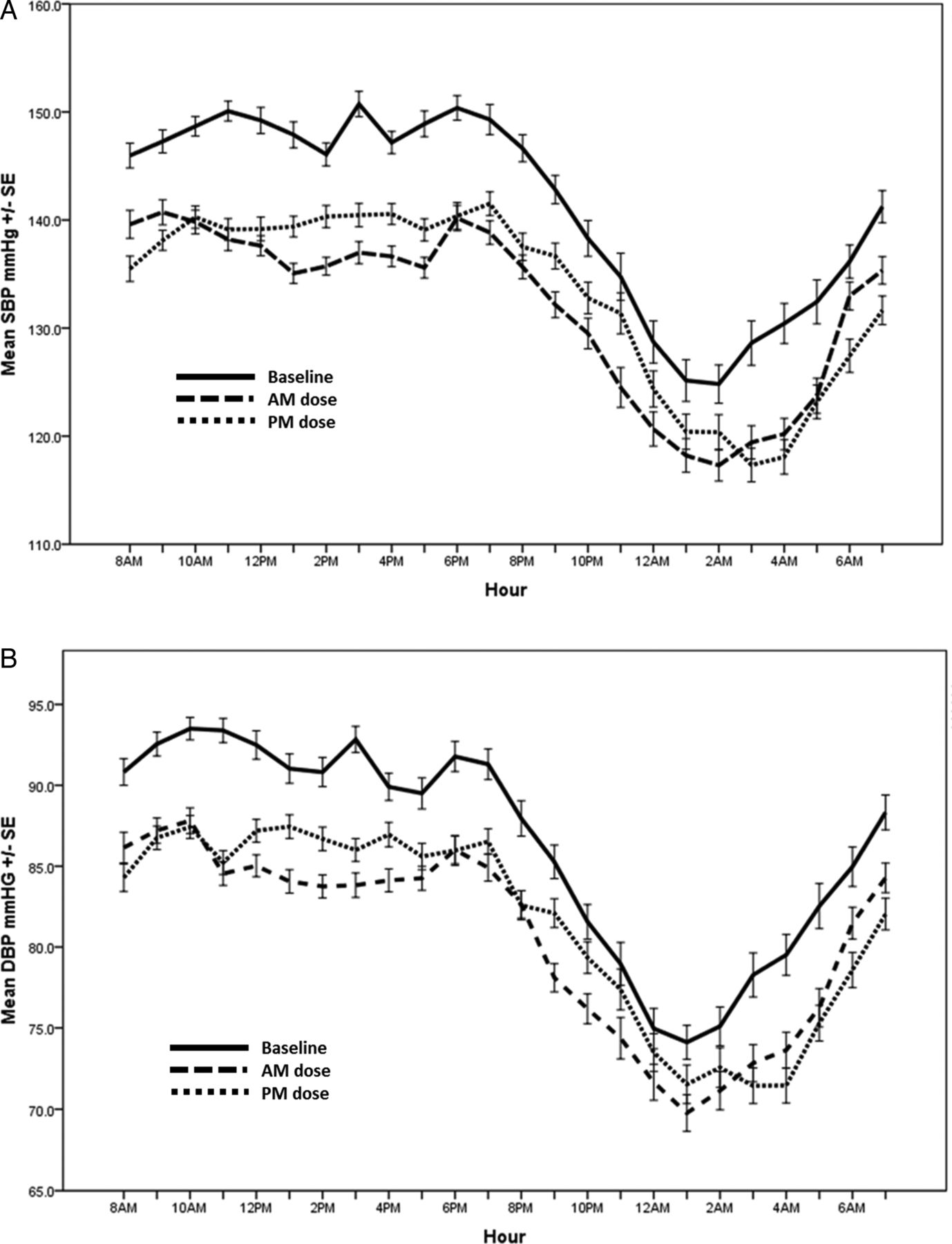
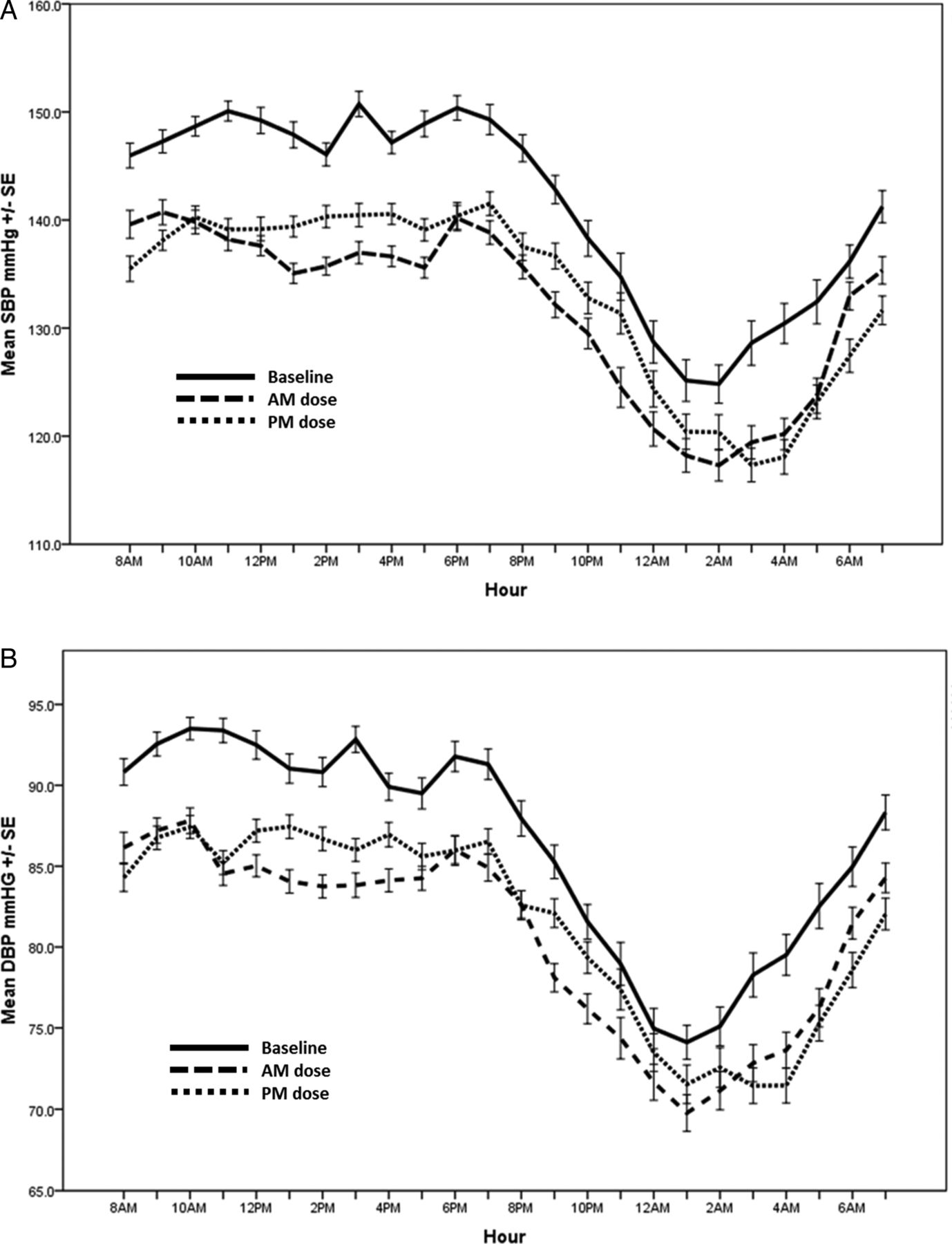
Our primary outcome of sleep SBP reduced significantly from baseline with the addition of perindopril regardless of dosing time (table 2 and figure 2). However, there was no significant difference in the reduction from baseline between the two dosing times (morning dosing reduced sleep SBP by 1.1 mm Hg more than evening dosing; p=0.14). In contrast, wake SBP reduced significantly more with morning dosing by 1.8 mm Hg (p<0.001).
Ambulatory blood pressure results before and after treatment with morning and evening dosing of perindopril*
{kind=link}
{kind=link}
Ambulatory blood pressure profile for all patients at baseline and after 6 weeks of morning (AM) dosing and evening (PM) dosing of perindopril. The top panel shows systolic blood pressure (SBP) over 24 hours and the bottom panel shows diastolic blood pressure (DBP) over the 24-hour period. I bars represent SE.
Similarly, sleep DBP reduced significantly with both evening and morning dosing without a superior effect with evening dosing. Morning dosing improved wake DBP more than evening dosing. The order in which patients were randomised to the medications had no significant effect on any BP result.
Office SBP reduced significantly but similarly from baseline with both morning and evening dosing. In contrast, office DBP reduced slightly more with morning dosing (by 1.9 mm Hg, p=0.046). There was no further change with the addition of CPAP (see online supplementary tables E1 and E2).
Summary statistics for the open-label CPAP phase are shown in table 3 (and online supplementary figure E1). Compliance data were available in all patients who completed the trial. The addition of CPAP reduced sleep SBP further (morning dosing: −3.3 mm Hg, p<0.001; evening dosing −3.2 mm Hg, p<0.001). However, the improvement with add-on CPAP was not different between the two dosing times for any BP parameter. CPAP compliance was 3:40 hours in the morning dosing group (n=40) and 3:54 hours in the evening dosing group (n=38). In patients who used CPAP for >1 hour a night (n=66), residual AHI as calculated by the device was dramatically reduced (6.8 events/hour±4.9 (SD)). Among these patients, compliance was 4:20 hours±1:53 in the morning dosing group (n=33) and 4:27 hours±1:40 in the evening dosing group (n=33); however, there was no difference in BP reduction between the two groups (results not shown).
Ambulatory blood pressure results before and after addition of CPAP to either morning or evening dosing of perindopril*
Non-dipping rates did not improve from baseline with perindopril regardless of dosing time (baseline: 24.7%, morning dosing: 28.7%, evening dosing: 29.1%, p values for all comparisons >0.7). Non-dipping rates also did not improve with either dosing time after the addition of CPAP (morning dosing with CPAP 27.5%, evening dosing with CPAP 18.4%, p=0.42).
In a subgroup analysis of non-dippers at baseline, evening dosing tended to convert more patients to a dipping BP profile (morning dosing 71.4% non-dippers, evening dosing 57.1% non-dippers), but this difference was not statistically significant (p=0.7). On the other hand, all other BP parameters were lower, and hence superior, in the morning dosing group (see online supplementary table E3).
The most common side effects were cough and dizziness. Of the 26 that reported a cough, 11 occurrences were thought to be related to the study medication and 15 were thought to be ‘possibly’ related to the medication. Only one patient withdrew prerandomisation due to cough, and two patients withdrew for the same reason after randomisation. Transient dizziness was reported in seven patients.
Medication compliance determined by pill counts during phase I and II exceeded 90% for both active and placebo tablets and for both morning and evening dosing times, although patients tended to be more compliant with morning tablets (see online supplement for details). During the CPAP phase, compliance was 95% for both dosing times.
Discussion
This study shows that in patients with moderate-to-severe untreated hypoxaemic OSA and stage I–II hypertension, evening administration of the antihypertensive perindopril did not produce a superior reduction of nocturnal BP as compared with morning administration. On the other hand, morning dosing was more effective at reducing daytime BP than evening dosing. The addition of CPAP to both dosing times further lowered sleep SBP, but evening dosing was not superior.
Although there was no chronotherapeutic benefit of evening dosing with the addition of CPAP, our results clearly showed that CPAP resulted in a significant reduction in sleep SBP >3 mm Hg. Even with this modest effect, epidemiological studies have shown that a similar (4 mm Hg) reduction in night-time BP is associated with a 10% reduction in stroke and 22% reduction in coronary heart disease among subjects aged <60 years.33 This additive effect of CPAP in improving BP has been demonstrated in other studies,10 ,12 ,34 ,35 although they have not all shown improvements in sleep SBP—the most important prognostic marker of cardiovascular disease.17
Appreciable chronotherapeutic effects on sleep BP have been reported in randomised controlled trials in patients with essential hypertension.36 These effects have also been emulated in an observational crossover chronotherapy study in patients with OSA with a finding that evening dosing of valsartan or the combination of valsartan and amlodipine as compared with morning dosing further reduced night-time SBP by 4.4±8.6 mm Hg (SD) and improved nocturnal dipping from 24% to 61%, as compared with 34% with morning dosing.29 This benefit far exceeds any effect from CPAP. Importantly however, this study was not randomised, placebo-controlled or blinded29 and when tested using these more robust trial design features in the current study, we did not find similar chronotherapeutic effects.
We chose to use perindopril as our antihypertensive agent for several reasons. First, it is recommended as one of the first-line antihypertensives for the treatment of hypertension in non-black individuals, which were the most prevalent race in our sample.37 In contrast, sympatholytic agents such as β-blockers are not recommended as an initial antihypertensive because they have been linked with poor outcomes.38 Currently, there is inadequate evidence to recommend any one particular class of antihypertensive as first line in patients with OSA and hypertension.39 ,40 Second, perindopril is a commonly prescribed, widely available medication with a safe side-effect profile.41 Third, ACE inhibitors and ARBs have been the most extensively studied classes of medications in positive chronotherapy studies. It could be argued that the lack of chronotherapeutic effect that we observed with perindopril may have been due to the long half-life (20–30 hours) of its active metabolite, perindoprilat.42 However, other ACE inhibitors and ARBs with long half-lives (ranging from 24 to 35 hours) have been studied in clinical chronotherapy studies and have shown a greater reduction in sleep BP with night-time than with daytime administration.43 This is thought to be due to the variability in pharmacokinetics and pharmacodynamics of medications across the 24-hour day as a consequence of circadian variation in internal processes,44–46 including the activity of the renin-angiotensin system.47
We suspect that one of the reasons our results were not as we had expected is that the typical circadian variation normally determining the pharmacokinetics of perindopril is probably altered in OSA, thereby negating any potential chronotherapeutic effects. However, this area of research is yet to be explored and remains unproven. Alternatively, excessive nocturnal sympathetic activation due to OSA may have had overriding hypertensive effects that negated chronotherapeutic effects. This would also explain the lack of improvement in dipping rates regardless of ingestion time. In support of sympathetic activation, the β-blocker atenolol has been shown to reduce night-time BP by more than other common drug classes.48 However, it remains to be established whether chronotherapeutic benefits would occur with such sympatholytic agents. The similar reduction in sleep SBP between evening and morning dosing after CPAP in our study is also consistent with a reduction in sympathetic activity. Why CPAP then failed to unveil any chronotherapeutic advantage with evening dosing may be attributable to suboptimal CPAP compliance. Several studies have highlighted the importance of rapid eye movement (REM) sleep in the pathophysiology of hypertension in OSA.27 ,49 ,50 In our current study with relatively poor CPAP compliance, it is possible that persistent OSA during the latter half of REM-predominant sleep may have continued to mask any potential chronotherapeutic benefit with evening dosing. Ultimately, this open-label CPAP component of the study was exploratory and likely underpowered to detect chronotherapeutic effects.
This study has several potential limitations. We excluded patients with resistant hypertension because they are by definition treatment resistant51 and were deemed unlikely to benefit from chronotherapy. We excluded elderly individuals (aged >65 years) as the association between sleep apnoea and hypertension is less clear in this cohort52 ,53 and they are more likely to suffer from isolated systolic hypertension.54 These exclusions limit the generalisability of our findings. Second, in order to reduce the burden on patients and subsequent withdrawals, we performed 24-hour ABPM whereas some of the previous chronotherapy studies were able to employ 48-hour BP monitoring. However, 24-hour monitoring is considered the reference standard for the diagnosis of hypertension,55 and we were able to achieve a high rate of successful BP readings (>85%). Furthermore, the only other chronotherapy study in OSA which also employed 24-hour monitoring did find a positive chronotherapeutic effect.29 Third, the CPAP intervention was an open-label observational phase without a placebo control condition. Although this may affect the strength of the findings, the magnitude of antihypertensive effect with CPAP was in keeping with previous randomised trials.7 ,8 Lastly, while we did not reach our target sample size, albeit by only three patients, our results indicate that our study was well powered.
In our trial, the rate of non-dippers was markedly less than in other chronotherapy trials that have shown impressive chronotherapeutic effects favouring evening dosing.20 While this may explain the lack of a chronotherapeutic effect in our trial, our subanalysis of non-dippers (although underpowered) still failed to demonstrate any chronotherapeutic benefit.
In terms of study strengths (and in contrast to the CPAP open-label phase), the main crossover trial was the first antihypertensive chronotherapy trial to use placebo control and the first randomised placebo-controlled antihypertensive chronotherapy trial in patients with OSA. The use of placebo in this type of trial is crucial as it has been well established that placebo medications can reduce BP significantly.56 A recent review found that placebo resulted in a reduction in SBP of 5.92 mm Hg in 52 trials of non-resistant hypertensives, and a reduction of 8.76 mm Hg in eight trials of resistant hypertensives.57 Blinding is arguably even more important in a chronotherapy study as unblinded patients allocated to night-time dosing may expect greater benefit, whereas those allocated to morning dosing may expect less benefit when fully informed about the study. Given that all chronotherapy-hypertension randomised trials to date have been performed without placebo control and appropriate blinding, the evidence for its effectiveness is not entirely robust. Other strengths of our study were a more objective and reliable determination of sleep and wake times through the use of actigraphy (rather than relying completely on self-reported diaries) and the low rate of dropouts throughout the trial.
The sleep apnoea hypertensive population (which represents almost half of all hypertensive patients58) is known to have high rates of nocturnal hypertension, and would stand to benefit significantly from a simple strategy directly targeting nocturnal BP. Our trial provides robust evidence that does not support evening administration of antihypertensive medication—at least for ACE inhibitors. Given the relatively small documented antihypertensive effect of CPAP on BP, maximising the application of pharmacotherapy is essential. In this regard, the chronotherapeutic effect of other classes of antihypertensives remains to be explored in this population, with the crucial incorporation of placebo. Only then will we truly know whether OSA is a unique comorbidity that renders chronotherapy useless and/or whether chronotherapy is a useful strategy in other comorbidities.
Acknowledgments
The authors would like to thank Mrs Kirsty Dodds and Dr Philip Cheuk Shing Lee for assistance with the running of the trial, Ms Frances Clements for her role as CPAP therapist, A/Prof Delwyn Bartlett for assistance with actigraphy analysis and Mr Gunnar Unger for web-based database creation. We would like to express our gratitude to Dr David Joffe and Dr Peter Buchanan for their help with patient recruitment. We would like to thank the patients for their valuable time, without which the study would not have been completed. Yasmina Serinel affirms that everyone who contributed significantly to the work and who wished to be acknowledged has been listed.
References
Footnotes
Contributors CLP, RRG, HA and PAC were responsible for the conception and design of the study. YS and CLP were responsible for the acquisition of data. YS, BJY and KHW were responsible for medical management of patients in the trial. YS, KHW and CLP performed the statistical analyses. All authors contributed to the interpretation of data, drafting and revising of the article and final approval of article.
Funding This study was funded by the Australian National Health and Medical Research Council Grant number 632758. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication. We received support from the pharmaceutical company Servier, who supplied us with both the active and placebo perindopril medication used in this study. CLP received an Australian National Health and Medical Research Council Career Development Fellowship (#1061545) and the Sydney Medical School Foundation Chapman Fellowship, which contributed to the completion of this work. RRG received an Australian National Health and Medical Research Council People Support Scheme, Practitioner Fellowship (#1022730), which contributed to the completion of this work. YS received a postgraduate scholarship from the University of Sydney to complete this work.
Competing interests None declared.
Ethics approval Northern Sydney Human Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Airwaves election special