Article Text

Abstract
Background Molecular pathways that regulate alveolar development and adult repair represent potential therapeutic targets for emphysema. Signalling via retinoic acid (RA), derived from vitamin A, is required for mammalian alveologenesis, and exogenous RA can induce alveolar regeneration in rodents. Little is known about RA signalling in the human lung and its potential role in lung disease.
Objectives To examine regulation of human alveolar epithelial and endothelial repair by RA, and characterise RA signalling in human emphysema.
Methods The role of RA signalling in alveolar epithelial repair was investigated with a scratch assay using an alveolar cell line (A549) and primary human alveolar type 2 (AT2) cells from resected lung, and the role in angiogenesis using a tube formation assay with human lung microvascular endothelial cells (HLMVEC). Localisation of RA synthetic (RALDH-1) and degrading (cytochrome P450 subfamily 26 A1 (CYP26A1)) enzymes in human lung was determined by immunofluorescence. Regulation of RA pathway components was investigated in emphysematous and control human lung tissue by quantitative real-time PCR and Western analysis.
Results RA stimulated HLMVEC angiogenesis in vitro; this was partially reproduced with a RAR-α agonist. RA induced mRNA expression of vascular endothelial growth factor A (VEGFA) and VEGFR2. RA did not modulate AT2 repair. CYP26A1 protein was identified in human lung microvasculature, whereas RALDH-1 partially co-localised with vimentin-positive fibroblasts. CYP26A1 mRNA and protein were increased in emphysema.
Conclusions RA regulates lung microvascular angiogenesis; the endothelium produces CYP26A1 which is increased in emphysema, possibly leading to reduced RA availability. These data highlight a role for RA in maintenance of the human pulmonary microvascular endothelium.
- COPD ÀÜ Mechanisms
- Emphysema
Statistics from Altmetric.com
Key messages
What is the key question?
What is the role of retinoic acid (RA) signalling in adult human alveolar repair?
What is the bottom line?
Exogenous RA stimulates angiogenesis and induces expression of vascular endothelial growth factor A (VEGFA) and VEGFR2 mRNA in human lung microvascular endothelial cells. The RA catabolic enzyme cytochrome P450 subfamily 26 A1 (CYP26A1) localises to microvascular endothelium in human lung tissue, whereas the RA synthetic enzyme RALDH-1 localises to a distinct population of alveolar cells that includes vimentin-positive stromal cells. Expression of CYP26A1 is increased in emphysematous lung tissue. These data suggest endogenous RA signalling regulates alveolar maintenance in man via the pulmonary microvascular endothelium, and furthermore, dysregulated endothelial RA catabolism may contribute to chronic lung disease.
Why read on?
This study for the first time describes how endogenous RA may regulate lung cellular repair processes in man and identifies the pulmonary microvascular endothelium as a therapeutic target for human lung regeneration.
Introduction
The adult mammalian lung has a robust capacity to regenerate following various types of injury; recent evidence suggests this is, at least partially, conserved in man.1 ,2 Whilst there has been progress in understanding factors that influence lung function decline in COPD, the molecular and cellular biology of human lung repair remains poorly understood.3 Molecular pathways that regulate lung development may also be important for lung repair in adulthood,4 and emerging data implicate the critical developmental factor retinoic acid (RA). RA is a small lipophilic molecule derived from dietary vitamin A (retinol). Circulating retinol binds to cytoplasmic retinol binding proteins (CRBP-1 or CRBP-2), and is converted by sequential oxidation by alcohol dehydrogenases and retinaldehyde dehydrogenases (RALDH) into biologically active all-trans RA (ATRA).5 Cellular retinoic acid binding proteins (CRABP)-1 and CRABP-2 transport ATRA either to cytochrome P450 subfamily 26 enzymes (CYP26A1, B1 or C1) for degradation, or to cognate type 2 nuclear retinoic acid receptors (RAR-α, RAR-β and RAR-γ) and retinoid X receptors (RXR-α, RXR-β and RXR-γ). These interactions regulate the activity of a wide assortment of genes.6
RA signalling has well defined roles in development and regeneration of various organs across species.7 In the lung, RA signalling regulates the outgrowth of nascent lung buds from foregut endoderm in the earliest stages of development,8 and is required post-natally in mice for alveologenesis, as demonstrated by alveolar defects resulting from targeted RAR disruptions.9–11 In adult rodents, endogenous RA signalling is required for alveolar maintenance; vitamin A deficiency in adult rats caused alveolar destruction and loss of gas-exchanging surface area, hallmarks of emphysema.12 Exogenous RA administration modulated alveolar regeneration following pneumonectomy in adult rodents and dogs,13 ,14 and remarkably, both RA and RAR agonists induced alveolar regeneration in adult rodent models of emphysema.15–18
Exogenous retinoids regulate lung function in man; maternal retinol supplementation in areas of endemic vitamin A deficiency improved childhood lung function.19 Recent data have identified a novel RA pathway gene as being important in determining adult FVC.20 In adults, serum retinol correlated with lung function in healthy subjects,21 and in patients with COPD, reduced serum retinol preceding decline in lung function.22 The cellular and molecular biology underlying these observations is not currently understood. RA signalling defects have been described in fibroblasts isolated from human emphysematous lung tissue.23 However, very little is known about the role of endogenous RA signalling in resident cells of the alveolus and how this might contribute to alveolar homeostasis.
In this paper, we characterise RA signalling in distal human lung and examine the role of RA in primary human alveolar epithelial and endothelial cell processes likely to be important in lung repair or regeneration. We demonstrate pathway components mediating RA synthesis and breakdown are localised to specific cell populations and identify increased capacity for RA catabolism in emphysematous lung.
Methods
Tissue samples
All human lung tissue samples were obtained from the Biobank of the Respiratory Biomedical Research Unit (BRU), Royal Brompton and Harefield NHS Foundation Trust (ethics reference number 10/H0504/9). Control lung was obtained from patients undergoing surgical resection for suspected lung tumours and comprised histologically normal parenchymal tissue not directly involved in the excised lung cancer. Emphysematous lung comprised parenchymal tissue obtained from patients undergoing lung volume reduction surgery for severe radiological emphysema. Lung samples were confirmed as histologically normal (control) or emphysematous according to tissue sections derived from regions immediately adjacent to frozen samples used in this study by an independent histopathologist (see online supplementary material). Patient demographics were obtained from anonymised clinical records with full consent (table 1). Control and emphysema groups were matched for age and sex. To determine the overlap between pathological emphysema and the physiology of the control group, which included patients with non-obstructed lung disease and patients with COPD, statistical tests were performed on lung function parameters between groups. Patients with emphysema exhibited a trend towards higher pack years, and lower FVC (p=0.07) than control patients. Patients with emphysema had significantly lower total lung capacity (p=0.04), FEV1, % predicted (p=0.0002), FEV1/FVC ratio (p=0.0028), and transfer coefficient for uptake of CO, corrected (KCOc, p<0.0001) than control patients, as expected.
Patient demographics and pulmonary function values associated with samples used in this study
Supplementary material
Cells and reagents
Pharmacological agents were used to manipulate the RA pathway in cells in vitro. ATRA was used to stimulate RA signalling, having strong affinity for all RARs.24 For activation of specific RARs, AM580 (RAR-α agonist),25 BMS453 (RAR-β agonist)26 and CD1530 (RAR-γ agonist)27 were used. To inhibit RA signalling, the pan-RAR inverse agonist BMS493, which antagonises RA signalling by competitively stabilising the association of RAR/RXR heterodimers with transcriptional co-repressors, was used.28 All retinoids (all-trans retinoic acid, ATRA (Sigma-Aldrich, Poole, UK # R2625), BMS493 (Sigma-Aldrich, #B6688), AM580 (Sigma-Aldrich, #A884), BMS453 (Tocris Bioscience, Bristol, UK #3409), CD1530 (Tocris Bioscience #2554)) were protected from photodegradation and stored as stock solution in dimethyl sulfoxide at −20°C for a maximum of 1 month.
A549 cells were purchased from the European Collection of Cell Cultures (Porton Down, UK) and maintained in Dulbecco's modified eagles medium (DMEM) supplemented with glutamine (2 mM), 10% foetal bovine serum (FBS), and penicillin and streptomycin (100 U/mL). Human lung microvascular endothelial cells (HLMVEC, Lonza, Wokingham, UK #CC2527) were maintained in EGM2-MV2 media (Lonza #3202) and used between passages 3 and 8 with no change in morphology or baseline tube-forming capacity. Primary human alveolar type 2 cells (hAT2) were isolated from normal resected lung tissue by partial enzyme digestion using bovine pancreatic trypsin (Sigma-Aldrich T8003) and preferential adherence to plastic,29 and cultured in DCCM-1 media (React Scientific, Ayrshire, UK) supplemented with 10% neonatal calf serum. Cells were washed with HBSS 48 hours after isolation to remove non-adherent cells, and formed a visually confluent monolayer by 3–4 days after isolation. hAT2 were not passaged.
Treatment protocols
Bovine serum contains retinoids, thus cells were starved in serum-free medium prior to experiments to minimise baseline RA signalling activity prior to treatments.30 To determine target gene induction, confluent A549 cells were serum starved for 24 hours and treated with ATRA (1µM) dissolved in medium for the indicated times, or with BMS493 dissolved in medium at the indicated concentrations for 20 hours, prior to RNA extraction. Confluent HLMVEC were serum starved for 24 hours and treated with ATRA for 4 hours prior to RNA extraction. Incubation times for mRNA induction were chosen to align with the durations of the functional assays. Proliferation was determined in sub-confluent A549 cells by incubation with antibody specific for phospho-serine 10 H3 (Cell Signaling Technology, #9701), counterstained with DAPI for nuclear staining, and visualised using a wide-field fluorescent microscope. The percentage of mitotic cells relative to total nuclei was calculated with Image J.
Epithelial scratch assay
Confluent A549 cells or hAT2 were serum starved for 24 or 2 hours respectively, and a linear scratch was generated with a p200 pipette tip. Cells were incubated at 37°C with the indicated treatments dissolved media containing 10% serum. To determine whether serum-derived retinoids interfere with exogenous ATRA, wound healing experiments were conducted using A549 treated with ATRA in media supplemented with charcoal stripped serum, which is depleted of lipophilic molecules which may have biological activity (such as steroids, peptide hormones and retinoids).31 Digital images were acquired immediately and at 18 hours after scratch, and the area covered by migrating cells was calculated with ImageJ software and expressed in pixels.32
Matrigel-based angiogenesis assay
Non-toxic retinoid concentrations were determined in HLMVEC with WST-1 reagent (Roche, Mannheim, Germany #05015944001). HLMVEC were incubated at 37°C for 4 hours in culture media containing ATRA, AM580, BMS453 or CD1530 at the indicated concentrations prior to incubation with WST-1. The amount of reduced formazan in the supernatants was spectrophotometrically determined at 450 nm with a plate reader. Only non-toxic concentrations were used for angiogenesis experiments (see online supplementary material). For the angiogenesis assay, HLMVEC were serum starved for 24 hours, trypsinised and seeded at 25 500/cm2 onto solidified growth factor-reduced Matrigel (BD Biosciences, Oxford, UK) in angiogenesis μ-slides (Ibidi, Martinsried, Germany #81506) and incubated at 37°C with EGM2-MV2 with all supplements except serum and hydrocortisone, containing ATRA, AM580, BMS453 or CD1530 at the indicated concentrations. After 4 hours, digital images were acquired, and mean total tube length per field of view quantified with ImageJ software.33 Data are expressed as mean total tube length normalised to mean of control.
Quantitative real-time PCR
RNA was extracted directly from cells treated in six-well plates, or from homogenised whole lung tissue (approximately 30 mg), with RNeasy Mini Kit (Qiagen). Following this, 1 µg RNA was reverse transcribed and analysed by quantitative real-time (qRT)-PCR using specific primers (Applied Biosystems, Foster City, California, USA; details provided in the online supplementary material). Individual Taqman-based assays were used for RALDH-1, RALDH-2 and RALDH-3, CYP26A1, CYP26B1 and CYP26C1, RAR-α, RAR-β and RAR-γ, RXR-α, RXR-β and RXR-γ, cellular retinol binding protein (CRBP)-1 and CRBP-2, CRABP-1 and CRABP-2, lecithin retinol acyltransferase (LRAT), β glucuronidase (GUSB) and β-2 microglobulin (B2M). All pathway components were expressed in control and emphysematous lung (see online supplementary material). qRT-PCR reactions were performed in triplicate, and the mean used to calculate fold change in expression between control and emphysema samples according to the ΔΔCT method.34 Glyceraldehyde 3 phosphate dehydrogenase (GAPDH), which we previously found to be the most stable reference gene for analysis of A549 cells with qRT-PCR,35 was used as the reference gene for A549 gene expression experiments. GUSB was the most stable reference gene for analysis of whole lung tissue with qRT-PCR,35 and in preliminary experiments we found the mean of GUSB and B2M to be suitable for qRT-PCR analysis of whole lung tissue and HLMVEC. To demonstrate the presence of RAR transcripts in hAT2 or HLMVEC, custom designed primers were used in non-quantitative end-point PCR reactions (sequences in online supplementary material), and product size confirmed by electrophoresis on 3% agarose gel.
Western blot
Total protein was extracted from whole human lung tissue (approximately 30 mg) in Cell Lysis Buffer (Cell Signaling #9803) containing protease inhibitors; 20 µg protein was loaded into NuPage 4–12% Bis-Tris gel (Invitrogen, Carlsbad, California, USA) alongside a protein ladder. Following transfer to nitrocellulose membrane, Western blot was performed with commercially available antibodies: anti-CYP26A1 antibody at 1:500 dilution (Sigma-Aldrich C6498), and anti-β-actin antibody at 1:2000 dilution (Cell Signalling, Beverly, Massachusetts, USA, #4970), with HRP-conjugated anti-rabbit secondary antibody (Cell Signaling #7074) at 1:5000 in blocking buffer. Band intensity was quantified with ImageJ software.
Immunofluorescence
Whole human lung tissue obtained from resections was inflated with low-melt agarose (Sigma-Aldrich A2576), and a Compresstome VF-300 microtome (Precisionary Instruments Inc., San Jose, California, USA) used to generate tissue slices 400 µm thick. Following fixation in 4% paraformaldehyde, whole-mount immunofluorescence was performed using primary antibodies anti-CYP26A1 (1:100, Sigma-Aldrich C6498), anti-RALDH-1 (1:100, Sigma-Aldrich HPA002123), anti-PECAM-1 (1:100, Sigma-Aldrich P8590) or anti-vimentin (1:100, Abcam ab8069) followed by secondary antibodies Alexa Fluor 555 donkey anti-rabbit (1:500, Life Technologies, A31572) and Alexa Fluor 647 donkey anti-mouse A31571) with ProLong Gold Antifade containing DAPI (Life Technologies) as counterstain. Immunofluorescence was visualised using a Zeiss LSM-510 confocal microscope.
Statistical analysis
Data were analysed with GraphPad Prism V.6.0 (GraphPad Software Inc, San Diego, California, USA). Non-parametric analyses were used throughout: Mann-Whitney test for two groups, and Kruskal-Wallis test followed by Dunn's post-test for multiple groups.
Results
RA does not directly modulate human alveolar epithelial type 2 cell wound healing
Treatment of A549 cells with ATRA (1 µM) increased message at 4 and 20 hours for RAR-β (p<0.05, figure 1A), which contains an RA response element in its promoter region.36 Accordingly, concentration-dependent reduction in RAR-β was demonstrated in A549 cells with the pan-RAR inverse-agonist BMS493, with maximal response at 1 µM (p<0.05, figure 1B). Non-quantitative PCR demonstrated the presence of RAR-α, RAR-β and RAR-γ transcripts in isolated primary human AT2 cells (figure 1D). To examine the role of RA signalling in alveolar epithelial monolayer repair, scratch assays were performed in A549 and primary hAT2. Neither RA pathway stimulation with ATRA nor inhibition using BMS493 modulated A549 (figure 1E, F) or hAT2 (figure 1G,H) wound closure following a scratch compared with control (medium supplemented with 10% serum alone). ATRA did not modulate A549 wound closure when dissolved in media supplemented with charcoal-stripped serum, which is depleted of serum-derived lipids including retinoids (see online supplementary material). Furthermore, treatment of sub-confluent A549 cells with exogenous ATRA (10 µM) did not influence proliferation (figure 1C). These data suggest that RA signalling does not directly influence alveolar epithelial repair.

Modulating RA signalling has no effect on A549 or primary human alveolar type 2 cell wound closure. (A) Confluent A549 cells were incubated with DMSO vehicle (empty bars) or with ATRA (1 µM, filled bars) for 4 or 20 hours, RNA extracted and quantitative real-time (qRT)-PCR for RAR-β performed with expression normalised to GAPDH. Results expressed as mean±SEM. n=4 per group, *p<0.05, **p<0.05 compared with vehicle, Kruskal-Wallis with Dunn's post test. (B) A549 cells were incubated with vehicle or with BMS493 for 20 hours and qRT-PCR for RAR-β performed. Results expressed as mean±SEM. n=3 per group, *p<0.05, **p<0.05 compared with vehicle, Kruskal-Wallis with Dunn's post test. (C) Sub-confluent A549 cells were incubated with 10% FBS-supplemented medium either alone or containing ATRA (10 µM) for 18 hours, then immunofluorescence performed for phospho-histone H3 serine 10, and digital images acquired. Nuclei of mitotic cells were calculated in ImageJ as a percentage of total nuclei assessed with DAPI counterstain. n=2 per group. (D) RNA was extracted from primary human AT2 cells and non-quantitative RT-PCR performed for RAR-α, RAR-β and RAR-γ. Reaction products visualised on agarose gel alongside a 25 bp DNA ladder (L, bright band at 125 bp). Bands correspond to expected size of amplicons: RAR-α 97 bp, RAR-β 70 bp, RAR-γ 103 bp. Confluent A549 cells were scratched with a pipette tip and incubated with 10% FBS-supplemented medium either alone or containing (E) ATRA or (F) BMS493 for 18 hours. Identical experiments were performed with confluent isolated primary human alveolar type 2 (hAT2) cells (G and H), which were scratched and incubated with 10% FBS alone or containing (G) ATRA or (H) BMS493 for 18 hours. Epithelial closure was quantified using ImageJ. Data presented as mean±SEM, n=at least 5 per group. Kruskal-Wallis test with Dunn's post-test did not detect any significant differences. ATRA, all-trans retinoic acid; FBS, foetal bovine serum; RAR, retinoic acid receptor; GAPDH, glyceraldehyde 3 phosphate dehydrogenase; DAPI, 4′,6-diamidino-2-phenylindole.
Human lung microvascular cells express retinoic acid receptors and are transcriptionally responsive to RA
Non-quantitative PCR demonstrated the presence of RAR-α, RAR-β and RAR-γ transcripts in HLMVEC (figure 2A). Using qRT-PCR, concentration-dependent induction of RAR-β was demonstrated in HLMVEC with a maximal response at ATRA 10 µM (p<0.05, figure 2B).

Role of retinoic acid in human lung microvascular endothelial angiogenesis. (A) HLMVEC RNA was extracted and non-quantitative RT-PCR performed for RAR -α, RAR-β and RAR-γ. Reaction products visualised on agarose gel alongside a 25 bp DNA ladder (L, bright band at 125 bp). Bands correspond to expected size of amplicons. (B) HLMVEC were incubated with DMSO vehicle or with ATRA for 4 hours, RNA extracted and quantitative real-time PCR for RAR-β performed with expression normalised to mean of GUSB and B2M. Results expressed as mean±SEM. n=4 per group, * p<0.05 compared to vehicle, Kruskall-Wallis with Dunn's post test. (C) HLMVEC were seeded onto solidified Matrigel and incubated for 4 hours without (control) or with ATRA. Tube-like structures are visible (arrowheads), representative images shown, original magnification ×10. (D) Quantification of mean total tube length per field of view with ImageJ, expressed relative to control (Ct), as mean±SEM, n=4 per group. *p<0.05 compared with control, Kruskal-Wallis with Dunn's post test. ATRA, all-trans retinoic acid; RAR, retinoic acid receptor; HLMVEC, human lung microvascular endothelial cells; GUSB, β glucuronidase; B2M, β-2 microglobulin.
RA stimulates angiogenesis in HLMVEC
ATRA (10 µM) significantly increased mean total tube length per field of view in a Matrigel-based angiogenesis assay (p<0.05, figure 2C, D). This was partially reproduced with AM580 (RAR-α selective agonist), with ∼30% induction at 1 nM (p<0.05), which diminished with higher concentrations (figure 3A). Toxicity of all retinoids at the concentrations used was excluded with WST-1 (see online supplementary material). Thus, the reduced effect of AM580 at 100 nM and above could be explained by decreased specificity for RAR-α.37 No effect of BMS453 (RAR-β selective agonist) or CD1530 (RAR-γ selective agonist) on HLMVEC angiogenesis was observed (figure 3B, C).
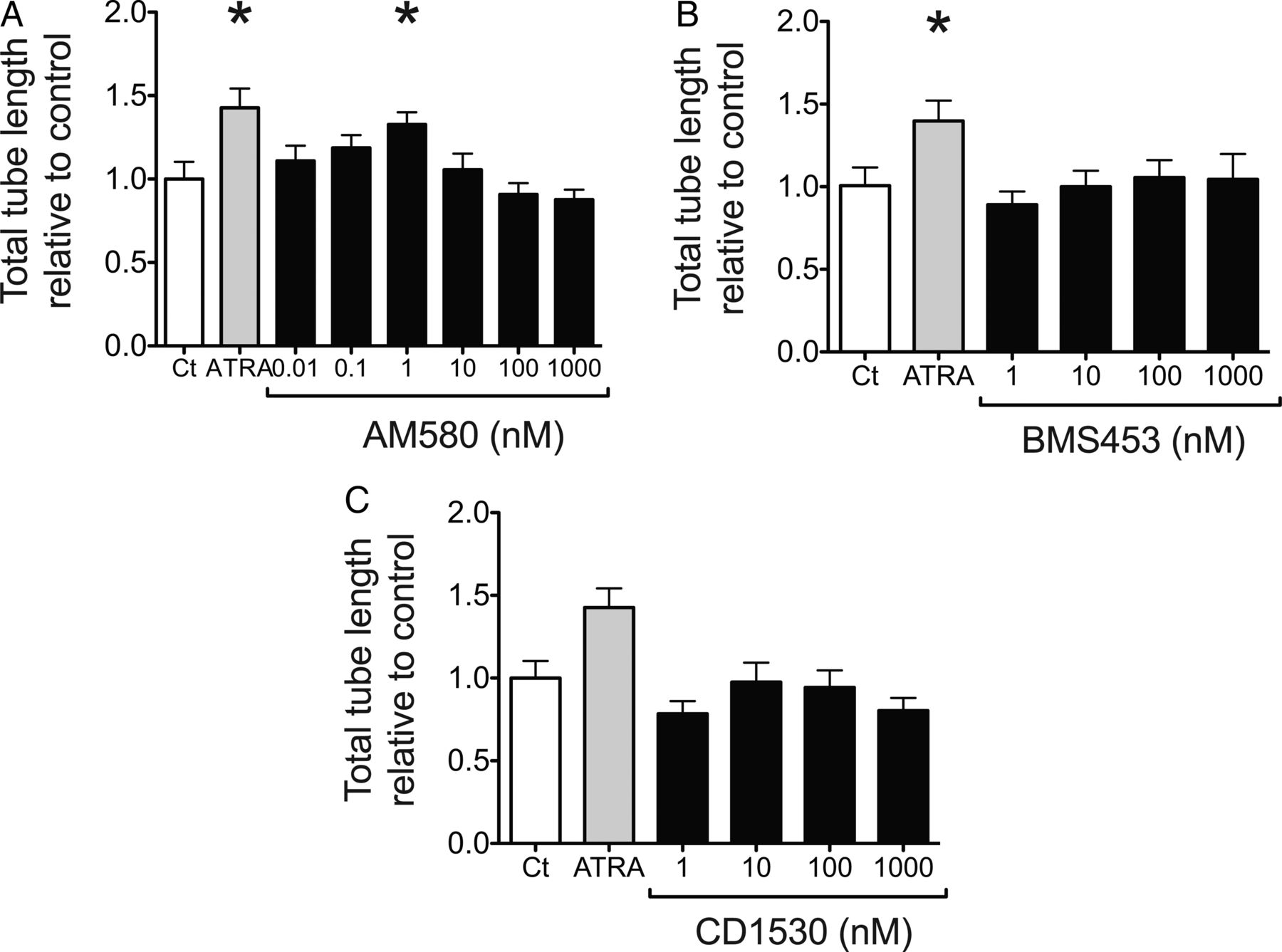
Role of RAR-β in HLMVEC angiogenesis. HLMVEC were seeded onto solidified Matrigel and incubated for 4 hours with control media (Ct), ATRA (10 µM) or with increasing concentrations of (A) AM580, (B) BMS453 or (C) CD1530, and mean total tube length per field of view quantified with ImageJ. Results expressed relative to control (Ct) as mean±SEM, n=at least 6 per group. *p<0.05 compared with control, Kruskal-Wallis with Dunn's post test. ATRA, all-trans retinoic acid; RAR, retinoic acid receptor; HLMVEC, human lung microvascular endothelial cells.
RA-induced HLMVEC angiogenesis is associated with induction of vascular endothelial growth factor pathway components
Significant induction of vascular endothelial growth factor A (VEGFA) mRNA was observed following 4 hours of incubation with ATRA (10 µM, p<0.05, figure 4A). A concentration-dependent induction of vascular endothelial growth factor receptor 2 (VEGFR2) was observed, with a maximal response at 10 µM (p<0.05, figure 4C). There was no effect on VEGFR1 expression (figure 4B).

Vascular endothelial growth factor signalling in retinoic acid-induced HLMVEC angiogenesis. HLMVEC were incubated with DMSO vehicle or ATRA for 4 hours, RNA extracted, and quantitative real-time PCR performed for (A) VEGFA, (B) VEGFR1 and (C) VEGFR2 with expression normalised to the mean of GUSB and B2M. Results expressed as mean±SEM, n=5 per group. *p<0.05 compared with control, Kruskal-Wallis with Dunn's post test. ATRA, all-trans retinoic acid; GUSB, β glucuronidase; B2M, β-2 microglobulin; VEGFA, vascular endothelial growth factor A; VEGFR1, vascular endothelial growth factor receptor 1; VEGFR2, vascular endothelial growth factor receptor 2.
RALDH-1 and CYP26A1 protein are present in distinct alveolar cell types
Whole-mount immunofluorescence on three-dimensional tissue slices derived from normal human lung parenchyma revealed positive immunostaining for the RA-degrading enzyme CYP26A1 in alveolar walls, co-localising with platelet endothelial cell adhesion molecule (PECAM)-1, a specific marker for endothelium (figure 5A–D), in a pattern resembling the microvascular capillary network. Positive immunostaining for the RA-synthesising enzyme RALDH-1 was partially co-localised with the mesenchymal cell marker vimentin (figure 5E–G). RALDH-1 and PECAM-1 exhibited little co-localisation (see online supplementary material). Secondary antibody specificity was demonstrated by absence of signal in sections incubated without primary antibody (see online supplementary material).
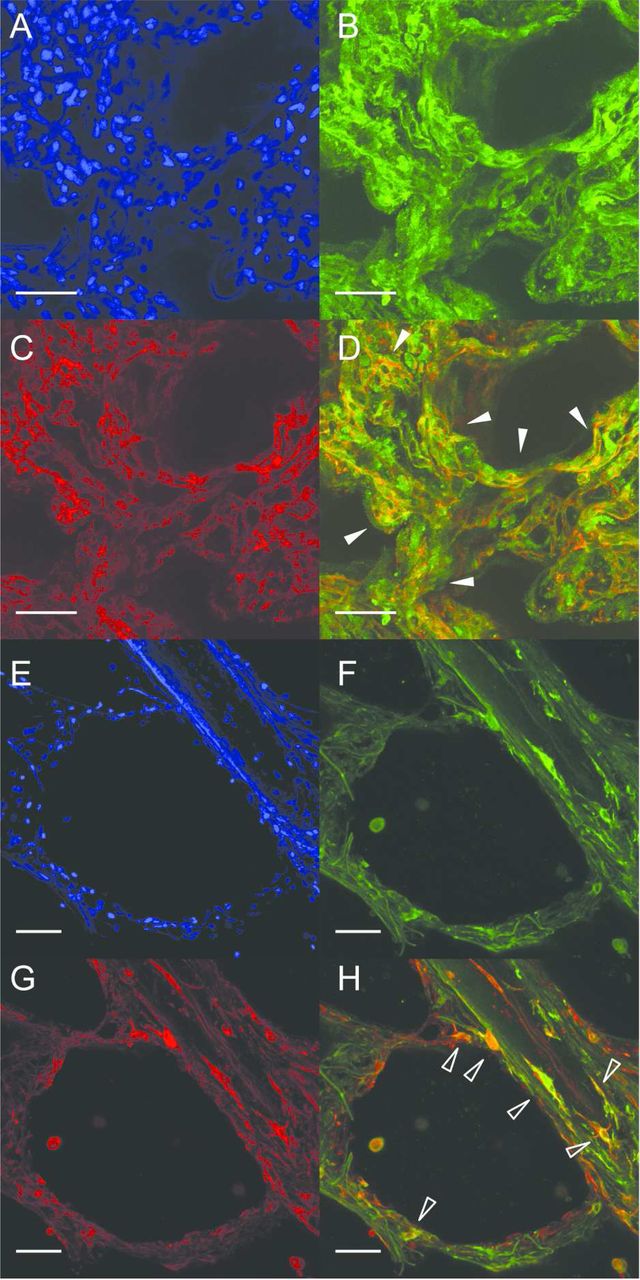
CYP26A1, RALDH-1, PECAM-1 and vimentin localisation in normal human lung parenchyma. Representative maximum intensity projections of Z-stacks through lung slices generated from histologically non-emphysematous adult human lung parenchyma, demonstrating localisation by whole-mount immunofluorescence of CYP26A1, RALDH-1, PECAM-1 and vimentin protein. Co-localisation of CYP26A1 protein (B, green) and PECAM-1 (C, red), to microvascular endothelial cells within the alveolar capillary network (merged in D, white arrowheads). Co-localisation of RALDH-1 protein (F, green) and vimentin (G, red), to a sub-set of stromal cells with fibroblast-like spindle morphology located near a small airway and within an alveolus (merged in H, empty arrowheads). RALDH-1 was also observed in occasional vimentin-negative cells (white arrowhead in H). DAPI was used as counter-stain (A and E, blue). 20× original magnification, scale bar=50 µm. CYP26A1, cytochrome P450 family 26 subfamily A polypeptide 1; PECAM-1, platelet endothelial cell adhesion molecule 1; RALDH-1, retinaldehyde dehydrogenase 1; DAPI, 4′,6-diamidino-2-phenylindole.
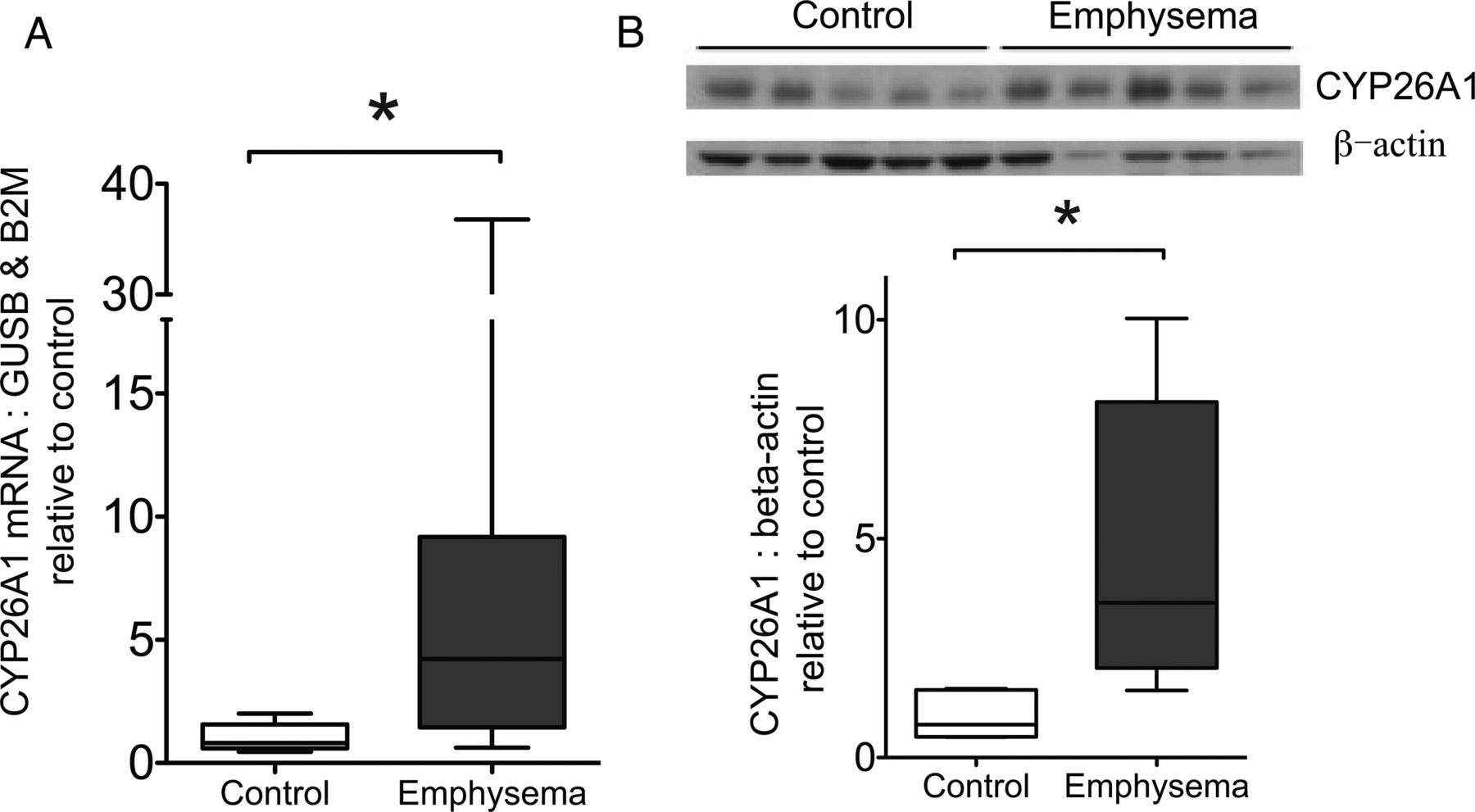
CYP26A1 mRNA and protein is increased in emphysematous lung tissue from patients with COPD
CYP26A1 mRNA expression was significantly higher in emphysematous tissue compared with control lung as shown by qRT-PCR (p=0.017, figure 6A). No other differences in expression were observed (see online supplementary material). By Western blot, relative to band intensity of β-actin loading control, immunostaining for CYP26A1 protein was significantly greater in emphysematous compared with control lung (p=0.016, figure 6B), showing an increase similar to mRNA levels.

CYP26A1 expression in control and emphysematous lung. (A) CYP26A1 mRNA expression in control (n=10) and emphysematous (n=9) lung. Tissue was processed for RNA extraction, and Taqman-based quantitative real-time PCR performed for CYP26A1 with expression normalised to the mean of GUSB and B2M. Results expressed relative to mean of control group. *p=0.017 Mann-Whitney test. (B) CYP26A1 protein was determined in whole lung tissue lysates from a separate cohort of patients (5 emphysematous, 5 control) using Western blot with anti-CYP26A1 antibody, with β-actin as loading control. Graph shows quantification of CYP26A1 band intensity, normalised to β-actin. Results expressed relative to mean of control group. *p=0.016, Mann-Whitney test. Data expressed as box and whisker plots with horizontal line representing the median, and upper and lower bounds of box representing IQR. GUSB, β glucuronidase; B2M, β-2 microglobulin; CYP26A1, cytochrome P450 family 26 subfamily A polypeptide 1.
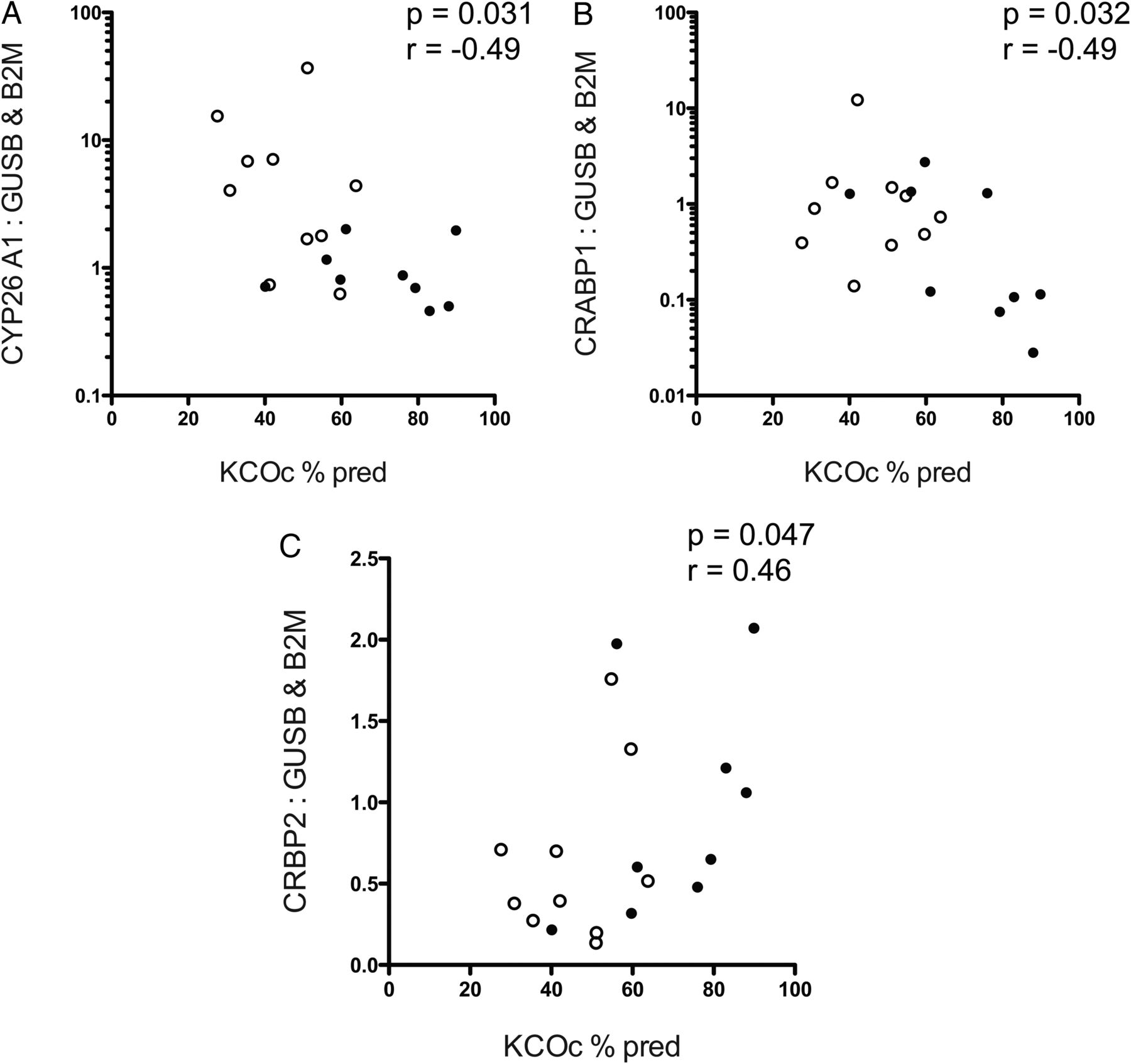
Reduced diffusion capacity correlates with RA pathway alterations consistent with diminished local RA availability
Significant negative correlations were observed between CYP26A1 and CRABP-1 expressions and KCOc % predicted (CYP26A1: p=0.031, r=−0.49, figure 7A; CRABP-1: p=0.032, r=−0.49, figure 7B, also see online supplementary material). A significant positive correlation was observed between CRBP-2 expression and KCOc % predicted (p=0.047, r=0.46, figure 7C). No correlations were observed between KCOc % predicted and expression of other RA pathway genes.

Correlations between CYP26A1, cellular retinoic acid binding protein 1, and cellular retinol binding protein 2 expression and transfer factor (KCOc % predicted) in COPD. Post-hoc statistical correlation of (A) CYP26A1, B) CRABP1 and C) CRBP2 mRNA expression normalised to mean of GUSB and B2M, with transfer coefficient for CO uptake (KCOc % predicted). For (A and B), Y-axes are log 10 scale for ease of display. Spearman's correlation, r=Spearman's coefficient. n=19, white circles=emphysema, filled circles=control. Abbreviations: GUSB, β-glucuronidase; B2M, β-2 microglobulin; CYP26A1, cytochrome P450 family 26 subfamily A polypeptide 1; CRABP1, cellular retinoic acid binding protein 1; CRBP2, cellular retinol binding protein 2.
Discussion
The RA signalling pathway controls development, maintenance and regeneration of diverse organ systems, across phylogeny.7 In the rodent lung, RA signalling has well characterised roles in regulating alveolar development.9–11 In adult animals, exogenous RA can modulate endogenous lung regeneration,13 ,14 and can induce alveolar regeneration in models of experimental emphysema.15–18 Regeneration of the alveolar-capillary membrane is critical for restoration of tissue integrity and gas exchange and requires coordinated growth of alveolar epithelium and microvascular endothelium. Alveolar regeneration following injury in animal models of disease involves proliferation and migration of AT2 cells across the basement membrane followed by differentiation into alveolar type 1 (AT1) cells.1 Here, we show that exogenous ATRA did not modulate AT2 monolayer scratch wound healing or proliferation, suggesting RA signalling is unlikely to directly control alveolar epithelial repair. In contrast, ATRA stimulated angiogenesis in HLMVEC, an effect partially reproduced by an RAR-α agonist, and ATRA induced mRNA expression of VEGFA and VEGFR2 in HLMVEC. We demonstrate that CYP26A1, which catabolises RA, was localised to the microvascular endothelium in human lung parenchyma, while the RA synthetic enzyme RALDH-1 partially localised to vimentin-positive fibroblasts. CYP26A1 mRNA and protein was increased in emphysematous lung tissue samples from patients with COPD compared with non-emphysematous control lung. These data suggest a mechanism by which endogenous RA signalling may regulate alveolar maintenance and repair in adult human lungs via the pulmonary microvascular endothelium. Furthermore, dysregulated endothelial RA catabolism may contribute to chronic lung disease by curtailing lung repair.
We demonstrate that the human lung epithelial cell line A549 is capable of responding to exogenous RA pathway modulation by altering expression of the RA target gene RAR-β (figure 1A,B). Furthermore, primary human AT2 cells express RAR-α, RAR-β and RAR-γ (figure 1C). However, neither RA pathway stimulation with ATRA nor inhibition with BMS493 affected A549 or hAT2 monolayer wound healing in a scratch assay (figure 1D–F, see online supplementary material), and ATRA did not influence proliferation of sub-confluent A549 cells (figure 1C). Together, this suggests that RA signalling does not directly regulate alveolar epithelial repair. This was surprising and contrasts with previous reports of a proliferative role of RA in rat neonatal alveolar epithelial cells,38 suggesting the role of RA signalling in juvenile and adult alveolar epithelium may be distinct. Therefore, although alveolar epithelial growth necessarily occurs during RA-induced adult lung regeneration, this may be indirect, mediated by effects of RA on other lung cell types.
We have demonstrated that HLMVEC isolated from adult lung express RAR-α, RAR-β and RAR-γ and are ATRA responsive. ATRA stimulated HLMVEC angiogenesis in vitro (figure 2A–D), suggesting that RA-mediated adult lung regeneration was in part mediated by microvascular angiogenesis. This is supported by prior reports: exogenous ATRA stimulated foetal sheep pulmonary artery endothelial cell (PAEC) proliferation and tube branching in vitro.39 RA was sufficient to protect pulmonary vascular development and thus maintain alveolarisation in neonatal mice following anti-angiogenic treatment,40 and in a canine model of post-pneumonectomy alveolar regeneration, RA administration preferentially increased microvascular capillary volume and the incidence of double capillaries, thereby enhancing alveolarisation.14
We performed exploratory experiments with pharmacological agonists to identify the specific RA receptors involved. Using the angiogenesis assay, we observed a ∼30% induction of tube formation by AM580 (10 nM), a RAR-α agonist (figure 3A), which may reflect a role for RAR-α in ATRA-induced HLMVEC angiogenesis. The lack of effect by RAR-β and RAR-γ agonists suggests neither receptor is involved in mediating ATRA-induced angiogenesis (figure 3B, C), however as BMS453 (RAR-β agonist) may antagonise RAR-α and RAR-γ in vitro, a role for RAR-β cannot be excluded.26 RAR-α agonists have previously been shown independently to induce angiogenesis,41 and lung regeneration.16 Notably, mice lacking RAR-α exhibited normal alveoli at 14 days, but failed to continue alveolar development between 14 and 50 days.9 This implicates RAR-α in a period of late alveolarisation, in which remodelling of the microvascular network is critical,42 supporting a role for RAR-α-mediated lung microvascular angiogenesis in postnatal alveolar growth and repair.
In further experiments to identify possible downstream signalling mediators of ATRA-induced HLMVEC angiogenesis, we measured mRNA expression of VEGF pathway members following ATRA incubation, and observed induction of VEGFA and VEGFR2 (figure 4A–C). VEGFA, a pro-angiogenic factor which mediates endothelial cell survival, permeability and angiogenesis through interaction with its receptors VEGFR1 and VEGFR2. It is thus possible that VEGF signalling mediates ATRA-induced HLMVEC angiogenesis. In support of this hypothesis, ATRA-induced foetal sheep PAEC branching was inhibited by a VEGF receptor antagonist,39 and RA-induced lung regeneration in mice was associated with increased VEGFR2 expression.43
We observed localisation of CYP26A1 protein to a sub-set of alveolar cells in normal human lung parenchyma, co-localising with PECAM-1 staining of the capillary network (figure 5A–D), indicating microvascular endothelial cells produce CYP26A1 protein. CYP26A1, CYP26B1 and CYP26C1 are highly specific, cytosolic cytochrome P450 enzymes that catalyse RA breakdown in RA target cells, and with RALDH-1, RALDH-2 and RALDH-3, regulate local RA availability.7 The human CYP26A1 gene contains three and a half conserved RA response elements in its 5′ promoter region and is highly inducible by RA.44 From our observation of CYP26A1 in pulmonary capillary network, we propose the endothelium as a principle target of RA signalling in the adult human lung. RALDH-1 protein partially co-localised with vimentin-positive stromal cells with fibroblast-like spindle morphology located around airways and within alveoli (figure 5E–G). RALDH-1, RALDH-2 and RALDH-3 catalyse the irreversible oxidation of the precursor retinaldehyde (retinal) into biologically active RA.7 RALDH-1 did not co-localise with PECAM-1, suggesting the endothelium is not a principle source of endogenous RA in the lung (see online supplementary material). Rather, our data suggest RA synthesis by lung stromal cells and a paracrine mechanism of RA signalling. Previous reports demonstrated RA synthesis and secretion by resident parenchymal lipid body-containing fibroblasts (lipofibroblasts) derived from rat lung.45 In mice during postnatal alveolar development, lipofibroblasts localise at the base of emerging septae, suggesting that these cells may drive alveolar-capillary growth.46 Our data are consistent with these findings, and correspond to RALDH-1 localisation previously observed in the postnatal mouse lung.47 RALDH-1 also localised to a sub-population of cells that were negative for vimentin, suggesting additional non-endothelial alveolar cell types might produce RA. Together, these data suggest the endothelium as a major target of paracrine RA signalling in human distal lung.
In our patient cohort, increased CYP26A1 mRNA and protein, and thus potentially increased capacity for RA catabolism, correlated with emphysema histology and physiology. CYP26A1 mRNA expression and protein were increased in emphysematous lung tissue from patients with COPD, compared with histologically normal lung tissue from patients with and without obstruction (figure 6A,B). Thus, increased CYP26A1 may be specific to emphysematous changes rather than to COPD generally. Accordingly, post-hoc analysis revealed a significant inverse correlation between CYP26A1 mRNA and KCOc % predicted (p=0.031, r=−0.49, figure 7A). CYP26A1 overexpression in HeLa cells in vitro increased RA degradation, rendering the cells insensitive to RA,48 and in vivo, low vitamin A status in mice increased susceptibility to developing emphysema from cigarette smoke exposure.49 We also observed a significant inverse correlation between CRABP-1 mRNA expression and KCOc % predicted (p=0.032, r=−0.49, figure 7B). CRABP-1 enhances RA degradation by transporting RA to CYP26s, and overexpression of CRABP-1 in F9 teratocarcinoma cells increased RA catabolism and diminished their sensitivity to RA.50 In addition, we found a significant positive correlation between CRBP-2 and KCOc % predicted (p=0.047, r=−0.49, figure 7C). CRBP-2 transports retinol to LRAT for conversion into retinyl esters,5 ,51 therefore, it is possible that decreased CRBP-2 in adult human lung could reduce formation of storage retinoids. Together, these data are consistent with the hypothesis that reduced local RA signalling is associated with the pathogenesis of emphysema.
We did not observe differences in CRABP-2 expression between emphysematous and control lung tissue samples, in contrast to a previous report of reduced CRABP-2 expression in lung fibroblasts from patients with emphysema.23 This discrepancy may reflect differences in using whole tissue for analysis compared with primary cells, which may be influenced by isolation protocols or culture conditions, although it is possible that in whole tissue samples, the transcriptional signature of specific cell types is diluted by the presence of other cell types. Because the main function of CRABP-2 is to facilitate RA signalling by transporting RA to the nucleus, these prior observations of decreased CRABP-2 expression would also result in reduced RA signalling,23 supporting our hypothesis that RA pathway dysregulation contributes to emphysema.
The hypothesis that increased capacity for RA catabolism contributes to emphysema suggests a pharmacokinetic explanation for the negative results of prior studies of exogenous RA in patients with COPD.52 ,53 Synthetic retinoids that are not susceptible to CYP26A1-mediated degradation thus may be more useful in this group of patients. Our finding that an RAR-α agonist partially reproduced ATRA-induced HLMVEC angiogenesis in vitro, together with prior reports that RAR-α agonists can induce lung regeneration in mice,16 suggests RAR-α may be a novel therapeutic target for patients with alveolar insufficiency.
Our study has limitations. Pharmacological agonists are not completely selective, the in vitro antagonism of RAR-α and RAR-γ by BMS453 being an example.26 To define further a role for specific RARs in HLMVEC angiogenesis, molecular disruption of RAR-α, RAR-β or RAR-γ, such as with siRNA-mediated knockdown or overexpression of dominant negative mutants, would be required. Similarly, to delineate a role for VEGF signalling, it would be necessary to demonstrate VEGFA or VEGFR2 protein induction by ATRA, and test if VEGF pathway inhibitors (such as the VEGFR2 inhibitor SU1498), or molecular disruption of VEGF pathway components, could interfere with ATRA-induced HLMVEC angiogenesis. Unbiased transcriptional profiling of HLMVEC following ATRA treatment may help reveal other downstream signalling pathways involved. Elucidating the mechanisms of ATRA-induced endothelial repair would help towards developing specific therapies that avoid off-target effects of modulating RA signalling, which has pleiotropic functions.
The scratch and Matrigel-based angiogenesis assays, although established and widely used models of cellular repair,32 ,33 cannot fully recapitulate the complex molecular events that occur during lung regeneration in vivo. As animal models often inadequately represent human physiology, complex human models of lung repair are needed. Recent progress with human organoid and lung-on-a-chip technology may offer exciting avenues for further testing the hypotheses outlined in our study.54 ,55 A multicellular assay such as co-culture of HLMVEC with lung fibroblasts may help to determine whether fibroblast-derived ATRA can induce HLMVEC angiogenesis, and further addition of hAT2 could test a role for indirect effects of RA on hAT2 repair via other cell types. Precision-cut lung slices are a promising alternative for defining the complex regulation of RA signalling between distinct alveolar cell types.56 Using immunofluorescence, we localised CYP26A1 and RALDH-1 to distinct alveolar populations, microvascular endothelium and fibroblasts respectively, suggesting a paracrine mechanism of RA signalling in human distal lung. This is strengthened by the lack of RALDH-1 expression in endothelium. However, we cannot exclude that endothelial cells express other RALDH subtypes (RALDH-2 or RALDH-3). Analysis of CYP26A1 levels in lung microvasculature of patients with emphysema would help to clarify whether endothelial RA catabolic capacity is specifically increased in emphysema. Additional studies of whether HLMVEC from patients with emphysema have altered capacity to respond to exogenous ATRA would provide a further test of our hypothesis.
In summary, we propose that RA is a key repair factor in the human alveolus (summarised in figure 8). Activation of RA signalling in microvascular endothelium by either endogenous RA or an exogenous retinoid stimulates angiogenesis, promoting alveolar regeneration (figure 8A). Conversely, degradation of endogenous RA by increased CYP26A1 impairs endothelial cell repair and may contribute to chronic lung disease (figure 8B). Together, these data contribute to our understanding of how a single essential fat-soluble nutrient, vitamin A, can directly modulate human lung responses in health and disease with potentially important clinical implications. The global variation in the prevalence of COPD is not fully explained by age, cigarette smoking and biomass smoke exposure, and other factors are therefore likely to be important.57 Vitamin A deficiency and subclinical deficiency are associated with poverty and are prevalent in the developing world. We suggest targeted intervention studies examining lung health in these populations might be informative.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model of role of RA signalling in emphysema and adult human lung regeneration. In this model, increased RA degradation, for example, through the CRABP-1–CYP26A1 axis, or decreased CRBP-2, contributes to decreased RA levels and a consequent deficiency in RA-mediated endothelial repair, which has a role in the pathogenesis of emphysema. In conditions of RA sufficiency or after exogenous RA administration, RA can stimulate microvascular angiogenesis, for example, through RAR-α activation, mediated by downstream pro-angiogenic signalling such as the VEGF pathway, thus promoting alveolar regeneration. CRABP-1, cellular retinoic acid binding protein 1; CRBP-2, cytoplasmic retinol binding protein 2; CYP26A1, cytochrome P450 subfamily 26 A1; RAR, retinoic acid receptor; VEGFA, vascular endothelial growth factor A; VEGFR2, vascular endothelial growth factor receptor 2.
References
Footnotes
CHD, UG, MJG and MH contributed equally to this paper
Contributors JPNB undertook acquisition, analysis and interpretation of data, and manuscript writing. JA undertook acquisition, analysis and interpretation of data for figure 5. MAM undertook acquisition and categorisation of lung tissue samples and provided images of tissue sections for online supplementary material. CHD, MJG, UG and MH contributed to the study design. All authors contributed to manuscript revision and final approval. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This project was funded and supported by the NIHR Respiratory Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.