Article Text

Abstract
Objective To assess whether FUT2 (secretor) genotype affects disease severity and airway infection in patients with non-cystic fibrosis bronchiectasis.
Participants Induced sputum samples were obtained from 112 adult patients with high-resolution CT scan-proven bronchiectasis and at least two exacerbations in the previous year, as part of an unrelated randomised control trial.
Outcome measures Presence of null FUT2 polymorphisms were determined by gene sequencing and verified by endobronchial biopsy histochemical staining. Outcome measures were FEV1% predicted, exacerbation frequency, and bacterial, fungal and viral components of the microbiota (measured by culture independent approaches).
Results Patients were grouped by FUT2 loss-of-function genotype; categorised as non-secretors (n=27, sese), heterozygous secretors (n=54, Sese) or homozygous secretors (n=31, SeSe). FEV1% was significantly lower in SeSe patients compared with sese patients (mean 61.6 (SD 20.0) vs 74.5 (18.0); p=0.023). Exacerbation frequency was significantly higher in SeSe (mean count 5.77) compared with sese (4.07; p=0.004) and Sese (4.63; p=0.026) genotypes. The time until first exacerbation was significantly shorter in SeSe compared with Sese (HR=0.571 (95% CI 0.343 to 0.950); p=0.031), with a similar trend for sese patients (HR=0.577 (0.311 to 1.07); p=0.081). sese had a significantly reduced frequency of Pseudomonas aeruginosa-dominated airway infection (8.7%) compared with Sese (31%; p=0.042) and SeSe (36%; p=0.035). In contrast, fungal, viral and non-dominant bacterial components of the microbiome were not significantly different between FUT2 genotypes.
Conclusions FUT2 genotype in patients with non-cystic fibrosis bronchiectasis was significantly associated with disease outcomes, with homozygous secretors exhibiting lower lung function, higher exacerbation number and a higher frequency of P. aeruginosa-dominated infection.
Trial registration number ACTRN12609000578202 (anzctr.org.au); Pre-results.
- Bronchiectasis
- Bacterial Infection
- Viral infection
Statistics from Altmetric.com
Key messages
What is the key question?
Does FUT2 genotype predict disease severity in non-cystic fibrosis bronchiectasis?
What is the bottom line?
Patients who had non-cystic fibrosis bronchiectasis with a homozygous polymorphism in the FUT2 gene have higher lung function, lower exacerbation frequency and lower prevalence of Pseudomonas aeruginosa-dominated airway infections, compared with patients without a FUT2 polymorphism.
Why read on?
The potential mechanisms by which FUT2 act provide insight into susceptibility to infection in chronic respiratory diseases, implicating FUT2 polymorphisms as an important marker to predict non-cystic fibrosis bronchiectasis disease progression.
Introduction
Non-cystic fibrosis bronchiectasis is a chronic airway disease characterised by irreversible bronchial dilation and persistent bacterial infections.1 Owing to its multifactorial aetiology, a patient's individual disease progression is difficult to predict.2 However, the composition of the airway microbiota, which differs substantially between patients, correlates with clinical markers of disease severity.3–5
Most notably, patients where Pseudomonas aeruginosa numerically dominates the composition of the airway microbiota have an accelerated decline in lung function, more frequent pulmonary exacerbations, greater sputum production and a greater requirement for antibiotic therapy.2 ,6–8 Respiratory viral9 and fungal10 infections have also been linked to bronchiectasis disease markers, which suggests that multiple infectious agents can contribute to the pathophysiology of non-cystic fibrosis bronchiectasis. While genetic loci in genes related to inflammation and airway remodelling have been previously investigated in non-cystic fibrosis bronchiectasis,11–15 little is known regarding common genetic polymorphisms that affect microbial acquisition in patients.
Variability in carbohydrate expression on mucosal surfaces is high, with some commensal and pathogenic microbes able to use glycans for adherence, induction of pathogenicity genes, and for use as carbon sources.16–18 Therefore, variability in glycan expression in the respiratory tract affects susceptibility to colonisation or infection with many bacteria, viruses and fungi.19 The FUT2 (secretor) gene encodes an α(1,2)fucosyltransferase, and homozygous loss-of-function mutations result in the inability to express ABH, Lewisb and Lewisy glycans on mucosal surfaces.20 Individuals who carry at least one functional copy of the FUT2 gene are known as ‘secretors’ (Sese or SeSe based on one or two functional copies, respectively) while those with two non-functional copies (approximately 20% of the Caucasian population) are known as ‘non-secretors’ (sese).20 ,21
Secretion of α(1,2)fucosylated glycans elicits a dichotomous effect on host–microbe interactions, the result of which is a difference in infection susceptibility, disease susceptibility and microbiome composition22 between secretors and non-secretors. For example, non-secretors have reduced incidence of influenza A, influenza B, rhinovirus and respiratory syncytial virus infections,23 but increased incidence of Neisseria meningitidis, Streptococcus pneumoniae24 and Candida albicans25 infections, compared with secretors. In relation to chronic respiratory conditions, non-secretor asthma patients present with fewer exacerbations26 and non-secretor cystic fibrosis patients (with severe impairment of lung function) have prolonged time until P. aeruginosa colonisation;27 however, non-secretors with COPD have a lower FEV1%.28 While one functional copy of FUT2 is sufficient to facilitate α(1,2)fucosylation, it is currently unclear whether heterozygote secretors (Sese) exhibit the same infection and disease susceptibility patterns as homozygote secretors (SeSe), or whether incomplete dominance presents, whereby heterozygotes display an intermediate phenotype.
We hypothesised that secretor genotype is a factor underlying variation in infection type and disease severity in non-cystic fibrosis bronchiectasis. To address this, we determined secretor genotype in a randomised controlled trail cohort of 112 adult patients with non-cystic fibrosis bronchiectasis, and performed secondary analysis by determining associations between genotype and key measures of disease severity. Additionally, we assessed whether secretor genotype was associated with changes in bacterial, viral and fungal components of the airway microbiota.
Methods
Study population
Patients with non-cystic fibrosis bronchiectasis were recruited as part of a randomised controlled trial designed to determine the efficacy of macrolides in preventing exacerbations.29 Adult patients aged 20–85 years with high-resolution CT scan-proven bronchiectasis, two or more exacerbations in the previous 12 months and daily sputum production were eligible. Patients were not receiving systemic corticosteroids, had not smoked cigarettes in the preceding 6 months, were macrolide naïve, and were not receiving nebulised antibiotics. Full details of inclusion and exclusion criteria for the study are detailed in online supplement E1.
Supplemental material
Clinical sample collection and processing
Induced sputum was collected at baseline as described previously30 and detailed in online supplement E2. Endobronchial biopsies were obtained from 32 patients who underwent bronchoscopies as an outpatient procedure. Biopsies were taken from subsegmental carinae of the lower lobes as detailed in online supplement E2.
Clinical measures of respiratory function and disease severity
Clinical assessments included FEV1, as a percentage of predicted value (FEV1%), Leicester Cough Questionnaire (LCQ) score, St. George's Respiratory Questionnaire (SGRQ) score, sputum weight over a period of 24 hours, physician defined pulmonary exacerbations (PDPEs) over the 48 weeks of the trial and pulmonary exacerbations in the prior 12 months (treated with either oral or intravenous antibiotics). During exacerbations, oral antibiotics were typically prescribed for milder exacerbations, where the patients were deemed well enough to return home, while intravenous antibiotics, in hospital, were prescribed if patients were deemed by the treating physician to have a severe exacerbation. Details of clinical data collection are detailed in online supplement E2.
All clinical measures analysed in this study were obtained prior to commencement of the clinical trial, except PDPEs which were recorded over the 48 weeks of the trial. Data relating to PDPEs were therefore presented for the total cohort and for those receiving placebo alone as a sensitivity analysis, in order to remove the possibility of confounding from macrolide use during the trial.
Secretor genotype and phenotype
Secretor genotype was determined by sequencing a 1162 bp region of the second exon of the FUT2 gene to identify loss-of-function polymorphisms. Sequences were aligned to a reference genome and cross-referenced with four known loss-of-function polymorphisms.20
Where bronchial biopsies were obtained, sections were stained for α(1,2)fucosylated glycans using horse radish peroxidase conjugated Ulex Europaeus lectin 1 (HRP-UEA1) (EY Laboratories, San Mateo, California, USA) to determine secretor phenotype, as previously described.26 A full description is detailed in online supplement E2.
Bacterial dominance
16S rRNA gene sequencing and identification of numerically dominant species (using specific PCR-based assays) were performed in 93 of the 112 patients, based on the availability of suitably archived samples, as described previously4 and provided in online supplement E2. Patients were categorised into one of three groups on the basis of numerically dominant bacterial species, which has been shown to be more clinically informative than culture-based methods.8 The groups comprise of P. aeruginosa-dominated, Haemophilus influenzae-dominated or dominated by a species other than these two. Previous quantification of the relative abundance of bacterial genera in this patient cohort has revealed that the airway microbiota is highly polarised in patients with H. influenzae-dominated and P. aeruginosa-dominated infections, with dominance of either exceeding 80% abundance.4 A summary of the dominant microbes in the patient cohort is provided in supplementary table E3. For analysis of the non-dominant microbiota, Pseudomonas or Haemophilus taxa was removed (where dominant) and the remaining relative abundance measures rescaled, as described previously.31
Fungal detection
Detection of C. albicans and A. fumigatus were performed by quantitative PCR in 78 patient samples, where suitably archived samples were available. Oligonucleotides and thermocycling conditions were performed as previously described.32 ,33 Detection limits were defined by serial dilutions of the clinical isolates (obtained from Flinders Medical Centre Microbiology Facility) and approximate copy numbers calculated (see online supplement E4).
Viral detection
Detection of nine common respiratory viruses (rhinovirus, adenovirus, influenza A, parainfluenza 1, 2, 3, respiratory syncytial virus, influenza B and human metapneumovirus) was assessed in baseline-induced sputum from the same 78 patients as those where fungal detection was performed. Detection was performed by the diagnostic virology unit, SA Pathology, using routine clinical diagnostic quantitative reverse-transcriptase PCR assays. Sample extraction was performed on a MagNA Pure 96 automated workstation (Roche Diagnostics, Castle Hill, New South Wales, Australia) and amplification and detection using specific in house primers and probes in validated PCR assays was performed on a LightCycler 480 with an extraction control and multiplexed assays with a maximum of three targets/reaction.
Statistical analysis
Clinical patient characteristics were tested for non-normality including skewness and kurtosis using the D'Agostino-Pearson omnibus test. Continuous data were analysed by one-way analysis of variance (ANOVA) with Tukey's post hoc test or by Kruskal-Wallis with Dunn's post hoc test, according to the distribution of the data. Ordinal data was analysed by Wilcoxon rank-sum test. Exacerbation counts were tested for Poisson distribution and subsequently analysed using a Wald test. Categorical data were analysed by χ2 or Fisher's exact test as appropriate. For time until first PDPE, Kaplan-Meier curves were produced and Cox proportional-hazards regression analyses were performed. All analyses were performed using SPSS (V.23.0, IBM, Armonk, New York, USA) or GraphPad Prism (V.6.05; GraphPad Software, San Diego, California, USA).
α diversity (Simpson's Index, Shannon-Weiner Index) and β diversity (Bray-Curtis similarity matrices) measures were calculated using sample-normalised, square root transformed relative operational taxonomic unit abundance using PRIMER (V.6.1.16; PRIMER-E, Plymouth, UK). Principal coordinate analysis was used to visualise clustering of samples based on their similarity matrices. The two-factor permutational multivariate analysis of variance (PERMANOVA) on the Bray-Curtis matrix was performed using PERMANOVA+package for PRIMER with 9999 random permutations.
Results
Secretor genotype and phenotype
FUT2 exon 2 genotype was determined in 112 patients, of whom 27 (24%) had a homozygous rs601338 428G→A base change (non-secretors), 54 (48%) were heterozygotes and 31 (28%) were homozygous for no mutation, termed sese, Sese and SeSe, respectively. The minor allelic frequency of this was 0.48 and the proportions fit Hardy-Weinberg distribution. Secretor phenotype, determined by histochemical staining of bronchial tissue, aligned with the genotype in all 32 patients with available biopsies (see online supplementary figure E5).
Clinical characteristics
Age, gender, duration of disease, drug treatments and comorbidities did not differ significantly between secretor genotypes (table 1). However, of the clinical measures of disease severity, homozygous secretor patients (SeSe) had significantly lower FEV1 (% predicted) compared with non-secretor patients (sese) (mean 61.6 (95% CI 54.3 to 69.0) vs 74.5 (67.3–81.6); p=0.023; table 1, see online supplementary figure E6).
Patient demographic and clinical characteristics
The number of exacerbations over the 12 month period prior to the trial intervention was also significantly higher in SeSe compared with sese (incident rate ratio (IRR)=1.42 (95% CI 1.11 to 1.78); p=0.004) and Sese (IRR=1.25 (1.03–1.51); p=0.026) patients (figure 1A). Of those, exacerbations requiring intravenous antibiotics (broadly indicative of a more severe pulmonary exacerbation) also differed, with no sese patients recording an exacerbation which required intravenous antibiotic therapy over the 12 months prior to the trial compared with 29% of SeSe patients having at least one exacerbation requiring intravenous antibiotics (figure 1B).
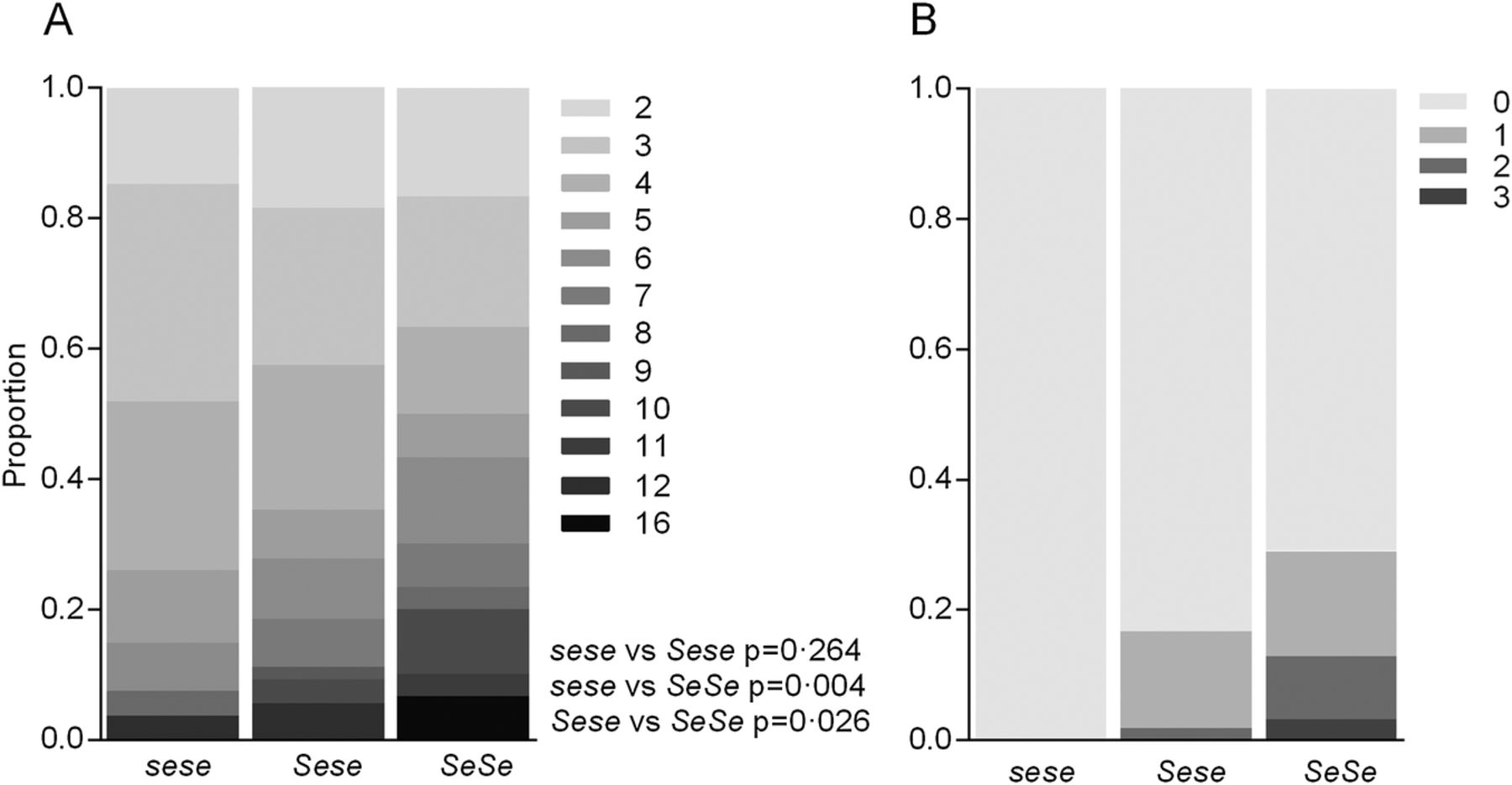
Effect of FUT2 genotype on 12 month pulmonary exacerbation count. Proportion of patients who had pulmonary exacerbations requiring any antibiotics (A) and which required intravenous antibiotics (B) over a 12-month period. Shading indicates number of exacerbations. p Values calculated by Wald test.
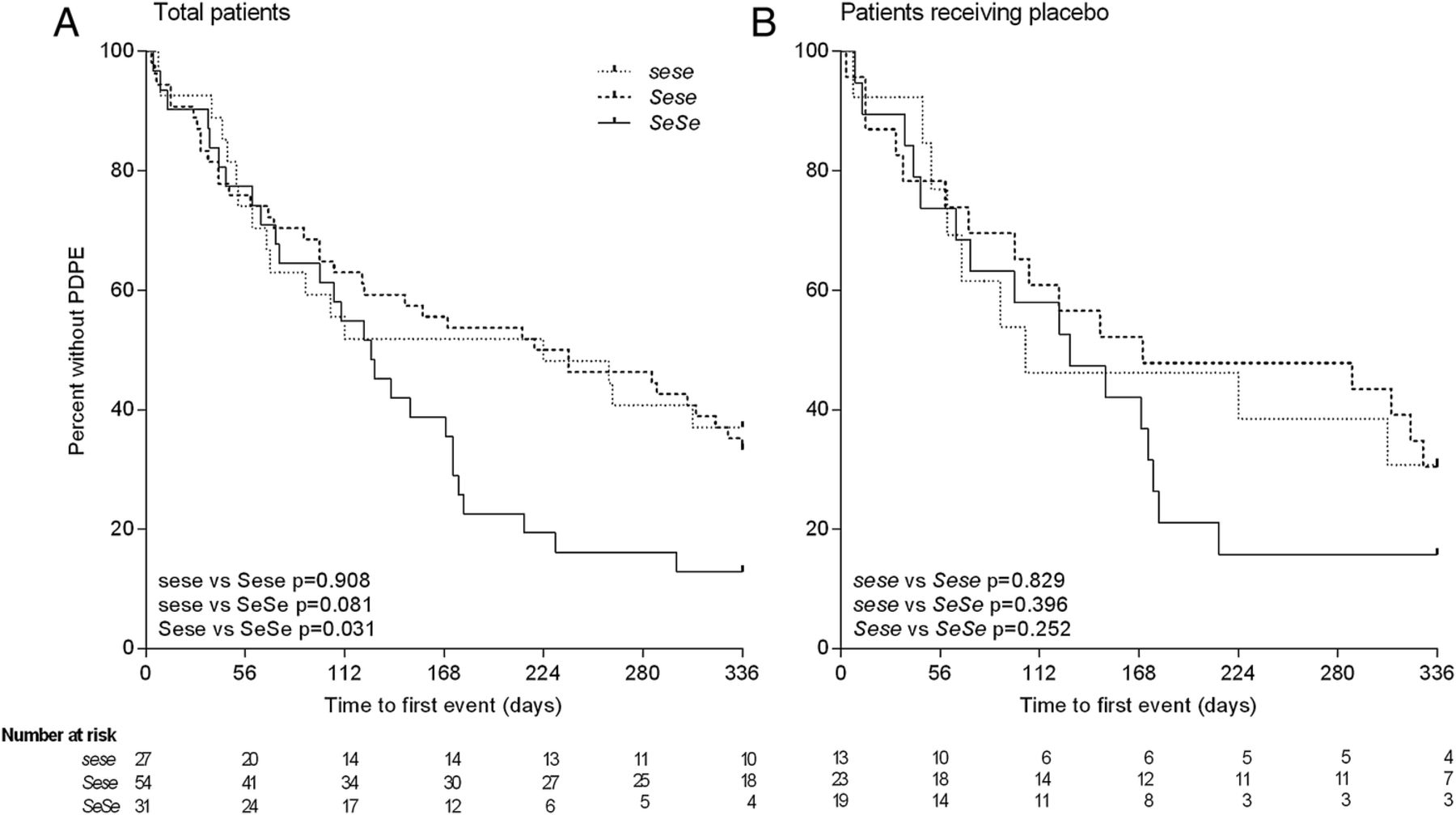
Over the course of the clinical trial, the primary outcome measures were the number of PDPE, as well as the time until the first PDPE. Significantly fewer sese patients recorded PDPEs compared with both Sese (IRR=1.52 (1.01–2.28); p=0.045) and SeSe (IRR=1.77 (1.15–2.72); p=0.009) patients (table 1 and see online supplementary figure E7). In the sensitivity analysis using the placebo subgroup alone, results followed the same trend (Sese IRR=1.76 (1.02–3.04); p=0.042 and SeSe IRR=1.69 (0.963–2.74); p=0.068, see online supplementary figure E7). The time until the first PDPE was also significantly longer in the Sese compared with SeSe genotype (HR=0.571 (95% CI 0.343 to 0.950); p=0.031), with a similar trend for sese patients compared with SeSe (HR=0.577 (0.311–1.07); p=0.081) (figure 2A). A similar pattern was also found in the placebo subgroup, however, due to the smaller sample size this did not reach statistical significance (Sese vs SeSe, HR=0.663 (0.329–1.34); p=0.252; sese vs SeSe, HR=0.669 (0.305–1.60); p=0.396, figure 2B). As many of the PDPEs were treated with antibiotic therapy, the total number of days on prescribed antibiotics was also tested, however, was not significantly lower in sese patients compared with Sese (p=0.502) or SeSe (p=0.094) patients (see online supplementary figure E8).

Effect of FUT2 genotype on time to first physician defined pulmonary exacerbation (PDPE). Total patients (A) and patients receiving only placebo (B). Kaplan-Meier curves are shown for the probability of remaining exacerbation-free according to FUT2 genotype. p Values calculated by Cox proportional-hazards regression model.
Airway bacterial predominance
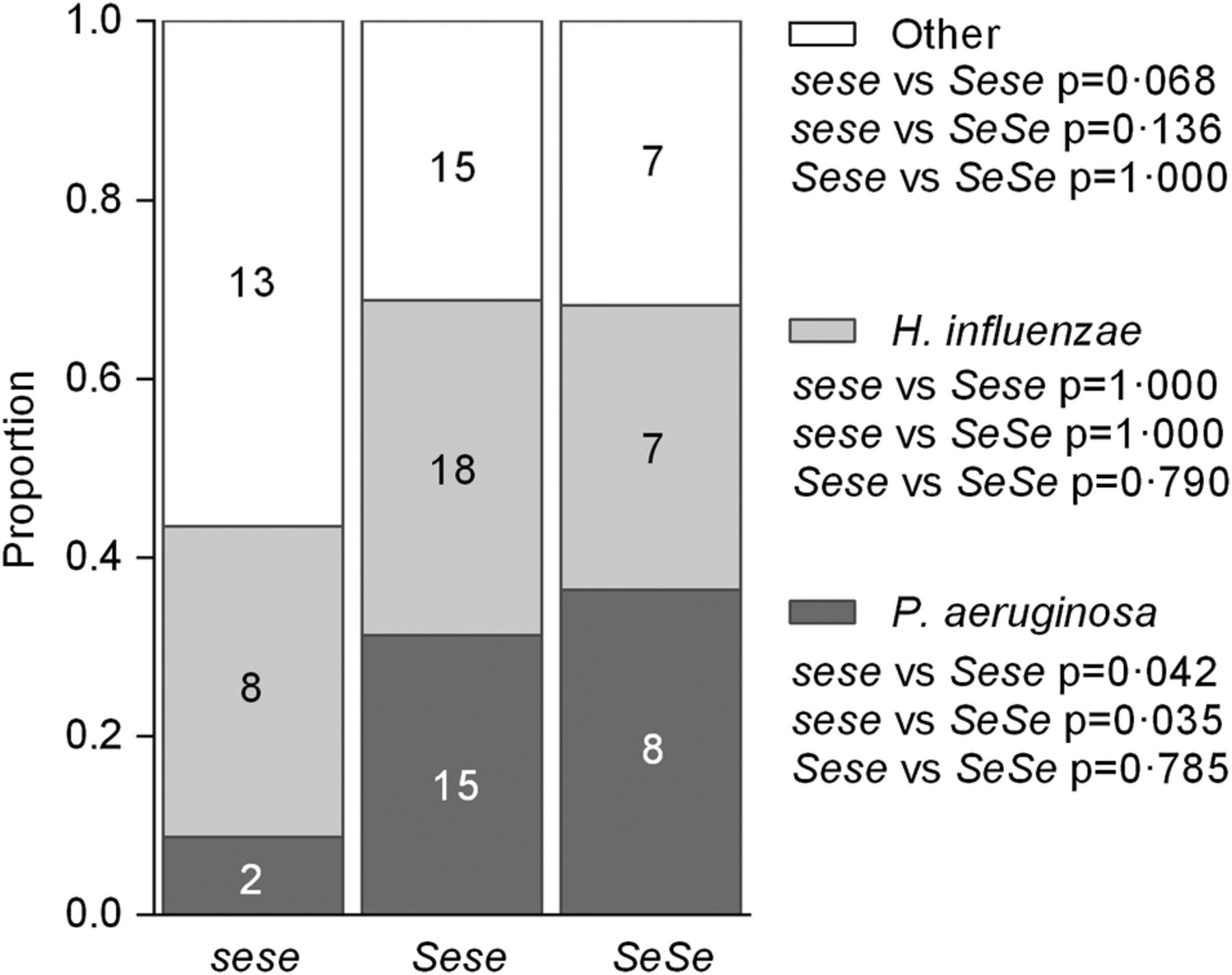
Of the 93 patients where airway bacterial composition was determined (23 sese, 48 Sese and 22 SeSe), 25 had infections dominated by P. aeruginosa, 33 by H. influenzae and 35 by any other species. The proportion of patients with P. aeruginosa-dominated infections was significantly lower in sese patients compared with Sese (p=0.042) and SeSe (p=0.035) patients (figure 3).
{kind=link}
{kind=link}
{kind=link}
Effect of FUT2 genotype on predominant airway infection. Proportion of patients who had airway infection dominated by Pseudomonas aeruginosa, Haemophilus influenzae or any other species. Numbers indicate number of patients. p Values calculated by Fisher's exact test.
Airway non-dominant bacterial taxa
To determine whether non-dominant components of the microbiota differed according to secretor genotype, differences in the non-dominant bacterial taxa, key fungal pathogens and key viral pathogens were compared. For non-dominant bacterial taxa, Shannon-Weiner Index and Simpson's Index were used to assess α diversity. Neither differed by secretor genotype (p=0.78 and p=0.73, respectively, see online supplementary figure E9). Principal coordinate analysis of Bray-Curtis distances revealed no distinct clustering between secretor genotypes (see online supplementary figure E10), which was confirmed by PERMANOVA test on the Bray-Curtis dissimilarity matrices (p=0.78), indicating that the composition of the non-dominant taxa did not significantly differ between patients based on FUT2 genotype.
Airway fungal and viral infection
Of the 78 patients where C. albicans and A. fumigatus were quantified (17 sese, 39 Sese and 22 SeSe), C. albicans was detected in 19/78 (24%) patients and A. fumigatus in 9/78 (11%) patients, although there was no difference in fungal presence between secretor genotypes (all p>0.05; see online supplementary figure E11). Similarly, of the nine viruses screened, only human rhinovirus was detected, in 4/78 (5%) patients, and there was no association with FUT2 genotype (χ2=1.5; p=0.48).
Discussion
To our knowledge, investigations of secretor status have not been previously performed in patients with non-cystic fibrosis bronchiectasis. In this cohort, the FUT2 G428A single nucleotide polymorphism (rs601338) allelic frequency was at 0.48. The rs601338 frequency reported in the healthy Caucasian population is 0.44;20 ,21 similar to the frequency reported in this cohort and suggesting that secretor status does not affect development of non-cystic fibrosis bronchiectasis. However, within non-cystic fibrosis bronchiectasis, we show that secretor genotype significantly affects disease severity.
By stratifying the non-cystic fibrosis bronchiectasis cohort by the presence of FUT2 null allele, we revealed that non-secretor patients (sese) had significantly higher lung function and lower frequency of pulmonary exacerbations, compared with homozygous secretor (SeSe) patients. Differences in patients' dominant airway microbiology (but not non-dominant microbiology, presence of common fungal species or detection of viral infections) were also found, with sese patients exhibiting significantly decreased frequency of P. aeruginosa-dominated airway infections.
Polymorphisms in the FUT2 gene are conserved at a high frequency in the population, likely driven by a dichotomous effect of α(1,2)fucosylated glycans on mucosal infection and disease susceptibility. In asthma for example, the frequency of a non-secretor phenotype is higher among Caucasian asthmatics compared with non-asthmatics,34 however, within patients with asthma, those with a secretor genotype are more prone to exacerbations,26 analogous to the findings of this study. In COPD, patients who are non-secretors have a lower FEV1% compared with secretors.28 Finally, in cystic fibrosis, no difference in lung function has been found between secretor and non-secretor patients, however, secretor patients in the ‘severe cystic fibrosis’ group have earlier onset of persistent P. aeruginosa infection compared with severe, non-secretor cystic fibrosis patients, which is consistent with our findings in non-cystic fibrosis bronchiectasis.27 It is also worth noting that along with chronic respiratory diseases, secretor status also significantly influences susceptibility to many gastrointestinal infections and diseases.17
The results of this study are also indicative of an incomplete dominance genetic model for secretor status. Patients with a heterozygous genotype displayed an intermediate phenotype in terms of lung function, total exacerbation frequency and frequency of exacerbations requiring intravenous antibiotics, despite Sese genotype considered equivalent to SeSe genotype. Previous findings also support incomplete dominance for FUT2, where similar, intermediate Sese genotype effects were found in relation to Crohn's disease gut microbiome and premature infant mortality.35 ,36 Further research is required to determine the effect of heterozygous versus homozygous secretor status on glycan expression and infection/disease susceptibility.
The precise mechanism by which secretor status affects infection and disease susceptibility is not entirely clear. It has been previously shown that even healthy sese individuals have a distinct intestinal microbiota composition from healthy Sese or SeSe individuals,22 linked to selective pressure from availability of different carbon sources.37 Secretor status could therefore affect infection and disease susceptibility via its effects on microbiome composition, as the microbiome has been shown to influence host immunity. Specific binding to α(1,2)fucosylated glycans has been implicated in the mechanisms of infection of several gastrointestinal pathogens (including Helicobacter pylori and Norwalk virus).18
In this cohort, we reported an increased frequency of P. aeruginosa-dominated infection in both Sese and SeSe patients with bronchiectasis. However, unlike Helicobacter pylori or Norwalk virus, P. aeruginosa does not encode any fucose catabolising genes and its fucose-specific adherence genes, PA-IIL and FliD, are not specific for α(1,2)fucosylated glycans,38 ,39 suggesting no direct link between secretor status and P. aeruginosa infection. Further detailed characterisation of P. aeruginosa growth, adherence and gene expression in sputum from individuals with sese and Sese/SeSe genotypes would be required to determine whether the effects of FUT2 provide any selective pressure. Other taxa which make up the respiratory microbiota could also be influenced by the presence of secretor glycans but were too subtle to detect by relative abundance analyses.
Alternatively, positive secretor status may affect severity of bronchiectasis via the well reported association with susceptibility to viral infections. Gastrointestinal viruses40 and respiratory viruses such as influenza A, influenza B, rhinovirus and respiratory syncytial virus23 are all more prevalent in individuals with a functional FUT2 gene. Respiratory viral infections are associated with pulmonary exacerbations in patients with non-cystic fibrosis bronchiectasis.9 Pulmonary exacerbations are characterised by inflammation, lung damage (which often permanently impairs lung function) and bacterial overgrowth, often requiring antibiotic therapy. A higher antibiotic burden has in turn been shown be selective for P. aeruginosa acquisition.3
We therefore propose a model whereby patients with a Sese or SeSe genotype have a greater susceptibility to respiratory viral infection, resulting in increased pulmonary exacerbation frequency, and leading to lower lung function, increased antibiotic exposure and subsequent selection for P. aeruginosa. Our results support this as secretors, particularly SeSe patients, had higher frequency of pulmonary exacerbations, lower FEV1%, and higher frequency of P. aeruginosa predominance, compared with sese patients. While results of our viral analysis showed a low prevalence of respiratory virus carriage across all groups, samples were taken from patients at a stable clinical baseline and are likely to differ in the period prior to or during exacerbations. Longitudinal viral detection at baseline and during exacerbations, would be required to adequately address this hypothesis and signifies a potential mechanism contributing to divergences in disease progression among patients with bronchiectasis.
We also assessed fungal colonisation in this patient cohort. Oral and vaginal carriage of Candida spp have been previously found to be higher in non-secretor individuals.25 ,41 Also, A. fumigatus encodes a lectin which preferentially binds α(1,2)fucosylated Lewis glycans,42 indicating secretor status may affect the presence of both C. albicans and A. fumigatus in bronchiectasis sputum. However, neither the presence of C. albicans nor A. fumigatus differed significantly based on secretor genotype, suggesting that fungal infections do not contribute to the difference in clinical symptoms reported between secretors and non-secretors.
Overall, the results of this study indicate that stratifying patients with non-cystic fibrosis bronchiectasis based on secretor genotype is likely to provide substantial prognostic value. Secretor status, which can be determined rapidly and at low cost, could be an important determinant of frequency of respiratory events, such as pulmonary exacerbations. Such information is relevant for clinical management, and also an important consideration in clinical trial design and the potential efficacy of vaccine strategies in different secretor groups.
There is a growing appreciation that moving towards precision medicine is a more effective approach to patient care, particularly for chronic disease management. Non-cystic fibrosis bronchiectasis has a complex underlying aetiology and implementing clinically informative details about patients' airway microbiology, inflammation, remodelling and respiratory physiology, as exemplified in this study, will ultimately be more beneficial for the development and application of more precise treatment options.
Acknowledgments
The authors dedicate this manuscript to the memory of Professor David Serisier, who passed away in May 2015, and whose loss will be deeply felt by the respiratory field. We would like to acknowledge Megan Martin for her role in obtaining the clinical samples, SA Pathology for performing viral quantification and Flinders Medical Centre Microbiology Facility for providing the fungal standards.
References
Footnotes
Contributors SLT, GBR, MAM and SW participated in the study's conception and design. SLT, GBR, ACHC and LDB participated in data collection. SLT and RJW were involved in statistical analyses. All authors participated in the interpretation of data and preparation and editing of the manuscript for intellectual content. All authors read and approved the final manuscript.
Competing interests None declared.
Ethics approval Mater Health Service Human Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial