Article Text

Abstract
Background Knowledge about the clinical spectrum of lung disease caused by variations in the ATP binding cassette subfamily A member 3 (ABCA3) gene is limited. Here we describe genotype-phenotype correlations in a European cohort.
Methods We retrospectively analysed baseline and outcome characteristics of 40 patients with two disease-causing ABCA3 mutations collected between 2001 and 2015.
Results Of 22 homozygous (15 male) and 18 compound heterozygous patients (3 male), 37 presented with neonatal respiratory distress syndrome as term babies. At follow-up, two major phenotypes are documented: patients with (1) early lethal mutations subdivided into (1a) dying within the first 6 months or (1b) before the age of 5 years, and (2) patients with prolonged survival into childhood, adolescence or adulthood. Patients with null/null mutations predicting complete ABCA3 deficiency died within the 1st weeks to months of life, while those with null/other or other/other mutations had a more variable presentation and outcome. Treatment with exogenous surfactant, systemic steroids, hydroxychloroquine and whole lung lavages had apparent but many times transient effects in individual subjects.
Conclusions Overall long-term (>5 years) survival of subjects with two disease-causing ABCA3 mutations was <20%. Response to therapies needs to be ascertained in randomised controlled trials.
- ABCA3
- Paediatric interstitial lung disease
- Surfactant protein
Statistics from Altmetric.com
Key messages
What is the key question?
What is the phenotype of patients with two disease-causing mutations in the ATP-binding cassette subfamily A member 3 (ABCA3) surfactant lipid transporter gene and how does it correlate to genotype?
What is the bottom line?
In a cohort of 40 subjects three phenotypes were differentiated, patients with null/null mutations died before the age of 6 months, long-term (>5 years) survival in this cohort was <20% and response to treatment was uncertain.
Why read on?
Explore in-depth clinical, biochemical, radiological and histological characteristics of patients with pulmonary disease caused by ABCA3 mutations for a better understanding of this orphan disease.
Introduction
The ATP-binding cassette subfamily A member 3 (ABCA3) protein is a 1704 amino acid protein, belonging to the ABC transporter family,1 encoded by a single gene with 33 exons located on chromosome 16.2 ,3 ABCA3 is predominantly expressed in lung tissue,4 localised to the membranes of the lamellar bodies of alveolar type II cells, where it is critical for pulmonary surfactant synthesis and processing.5 ABCA3 mutations have been associated with lethal neonatal respiratory distress,6–8 paediatric and adult interstitial lung disease,8–12 and surfactant metabolism dysfunction.13 ,14 In a recent study, genotype-phenotype correlation with respect to outcome was analysed in a cohort of 185 patients.7 With respect to clinical details, several case reports6 ,15–20 and one case series with nine patients are available,21 larger studies reporting clinical phenotype details with association to genotype are lacking.
In this study, we broaden the clinical knowledge about this patient group by presenting genetic, biochemical, clinical, radiological and histological findings in a cohort of 40 patients from Europe.
Methods
Patients
All children and adults identified in the programme for rare lung diseases of the Kids Lung Register (KLR) between 1 January 2001 and 30 June 2015 with homozygous or compound heterozygous mutations in the ABCA3 gene were included in this study (figure 1). Previously published patients were included with additional information and/or for completion of the cohort (see online supplementary tables S1 and S2). The patients' status at last follow-up was compared with the initial presentation and was categorised as dead, sick-worse (the patient had more severe symptoms), sick-same (the symptoms were of similar severity) or sick-better (the patient had milder symptoms and/or less therapy). The patients' response to therapies was analysed from the records and classified in comparison to before the therapy was started: no improvement (therapy did not yield any positive effect), moderate improvement (therapy yielded some positive effect, eg, milder mechanical ventilation, less oxygen needed and/or effect was only transient) and good improvement (therapy yielded a clear positive effect, eg, weaning from mechanical ventilation possible).

Flow chart of patients included in the study. DPLD, diffuse parenchymal lung disease.
supplementary data
Genetic analysis
ABCA3 was analysed using Sanger sequencing (primers are indicated in online supplementary table S3). Variations were grouped according to Wambach et al:7 missense mutations, insertions and deletions were classified as ‘other’ mutations, whereas frameshift mutations, nonsense mutations or deletion of an entire exon were classified as ‘null’ mutations.
Structure-function analysis of previously undescribed ABCA3 mutations
The predicted effect of ABCA3 mutations on protein function was calculated using PROVEAN,22 SIFT23 and PolyPhen2.24
Total protein and surfactant protein B and C concentration in bronchoalveolar lavage fluid
Surfactant protein B (SP-B) and surfactant protein C (SP-C) were determined in bronchoalveolar lavage fluid (BALF), obtained prior to exogenous surfactant application, using quantitative western blotting.25 ,26
Lung biopsy, histopathology, electron microscopy and immunohistochemistry
All available lung biopsies were collected as paraffin blocks or slides and evaluated by a pathologist specialised in pulmonary diseases (SR). Histology was scored by severity (0=none, 1=discrete, 2=moderate, 3=strong).27 When material was not available, we used histopathological categorisation and result of electron microscopy in the records.
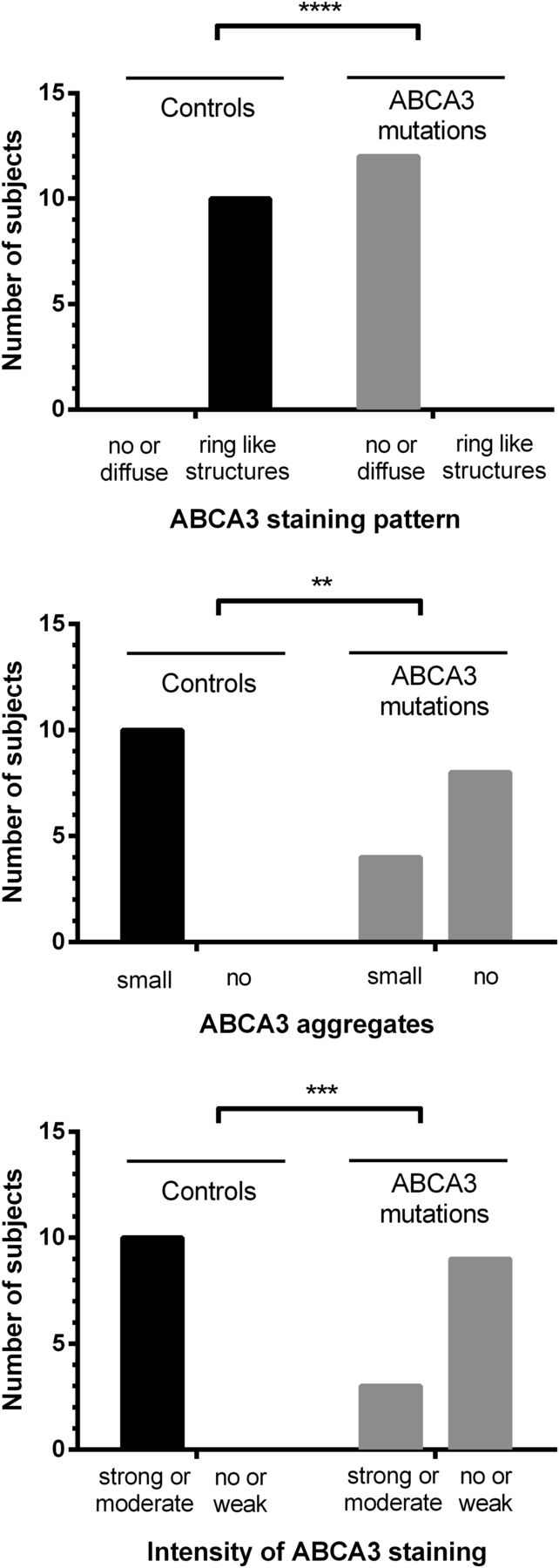
Immunohistochemical staining for ABCA3 was performed in all 12 available slides. After pretreatment with Target Unmasking Fluid (Panpath, Amsterdam, the Netherlands) at 90°C, primary antibody against ABCA3 protein (Seven Hills Bioreagents, Cincinnati, USA) was used for 60 min at room temperature, diluted 1:8000. Primary antibody was detected by Vectastain ABC-Kit Elite Universal (Vector Laboratories, Burlingame, USA) and AEC+ chromogen (Dako, Santa Clara, USA), counterstained with haematoxylin Gill's formula (Vector Laboratories). Microscopic pictures were taken with a Leica DM4000 (Leica, Wetzlar, Germany). Slides were blinded and scored semiquantitatively with respect to ABCA3 staining pattern (0=no staining, 1=diffuse staining, 2=ring-like structures stained), ABCA3 aggregates (0=no aggregates, 1=small aggregates, 2=big aggregates) and intensity of ABCA3 staining (0=no staining, 1=weak staining, 2=moderate staining, 3=strong staining). Scoring was analysed using Fisher's exact test. As controls we used tumour-free lung tissue from children (n=10, median age 9.3 years (2.3–17.5)), who received biopsy for diagnosis of pulmonary nodules associated with oncological diseases (nephroblastoma (n=6), hepatoblastoma (n=1), osteosarcoma (n=2), nerve sheath tumour (n=1)).
Radiological studies
Radiological studies were scored by a paediatric radiologist (VT) according to the criteria of Fleischner Society.28 CT scans and chest X-rays (CXR) were rated in three sections on the left and right sides: apex to carina, carina to lower pulmonary vein and lower pulmonary vein to diaphragm. The incidence of hilar lymphadenopathy, ground glass opacity, cysts, bronchial wall thickening, bronchiectasis/peripheral bronchial dilatation, reticular pattern, nodular pattern, lobe retraction and consolidation was evaluated for each lung section on both, CXR and CT scans. CT scans were in addition assessed for honeycombing, crazy paving and paraseptal emphysema, CXRs for hyperinflation. Each item was scored as 0=none, 1=discrete, 2=diffuse or 3=very strong. Mean values were calculated per patient for each feature.
Statistical analysis
Prism 6 for windows was used for statistical analysis. The Fisher's exact test was used to test the hypothesis that frequencies of ABCA3 staining pattern, ABCA3 aggregates and the intensity of ABCA3 staining were different between normal subjects and patients carrying ABCA3 mutations. Survival curves were compared by two methods: the log-rank test and the Gehan-Breslow-Wilcoxon test, which gives more weight at early time points.
Ethics
All parents or guardians of the children gave informed consent; adolescents assented. Retrospective data analysis was approved by the institutional review board (EK 026-06), and collection of data and material was performed under the project FP7-305653-chILD-EU (EK 111-13).
Results
Between 1 January 2001 and 30 June 2015, 1153 patients with diffuse parenchymal lung disease (DPLD) were registered in the KLR. The DNA of 242 of these patients was sequenced for ABCA3 mutations. Sixty-nine patients had at least one variation in the ABCA3 gene. Twenty-nine patients with only a single disease-causing mutation and/or patients with variations predicted not to be damaging were excluded from the study. Of 40 patients with two disease-causing ABCA3 mutations, 22 patients were homozygous and 18 patients compound heterozygous (figure 1).
Clinical course of patients with homozygous mutations
Twenty-two patients (15 male) carried homozygous ABCA3 mutations (see online supplementary table S1). The parents of 17/18 patients were consanguine. The parents of 14/17 patients were Caucasian, 2/17 African, 1/17 Indian. In 8/10 families with sufficient information one or two prior abortions, and/or family members who died from lung disease between 0 month and 11 months of age were recorded (see online supplementary table S4). Immediately or within a few hours after birth, 19/22 patients presented as mature newborns with severe respiratory distress, requiring mechanical ventilation. Three of the patients described in detail by Campo et al10 developed symptoms late during disease course. Hundred per cent of patients with null/null mutations died at a median age of 0.11 years, while 62% of patients with other/other mutations died at a median age of 0.25 years (see online supplementary table S1, figure 2). Two homozygous patients underwent lung transplantation (LTX), one is still alive at 3.3 years. Therapy with exogenous surfactant was applied to 17 patients, with no effect in 7 patients, and moderate but transient improvement in 9 patients. Therapy with systemic steroids was administered to 21 patients, with no effect in 14 patients, moderate improvement in 5 patients and good improvement in 1 patient. Therapy with hydroxychloroquine was tried in 10 patients, showing no effect in 5 patients, moderate but transient improvement in 3 patients and good improvement in 1 patient. Azithromycin application showed moderate improvement in one patient, and had no effect in two patients (see online supplementary table S1).

Kaplan-Meier curve of the survival of all patients with null/null and other/other or null/other mutations in the ATP-binding cassette subfamily A member 3 (ABCA3) gene (upper part) and same patients sorted according to homozygous (consanguinity likely) or compound heterozygous mutational status. Survival curves were compared by two methods: the log-rank test (upper panel p=0.0148; lower panel p=0.1629) and the Gehan-Breslow-Wilcoxon test, which gives more weight at early time points (upper panel p=0.0307; lower panel p=0.0041).
Clinical course of patients with compound heterozygous mutations
Eighteen patients had compound heterozygous ABCA3 mutations, 15 were female (see online supplementary table S2). The majority of the parents were heterozygous, not consanguine. Sixteen of 17 families were Caucasian, 1/17 African/Asian. Family history is summarised in online supplementary table S5. Age at presentation was more variable in patients with compound heterozygous ABCA3 mutations compared with patients with homozygous mutations. Thirteen patients presented with neonatal respiratory distress syndrome (RDS), three later in childhood and one in adulthood. Outcome in patients with compound heterozygous ABCA3 mutations was also variable: one patient with lung transplantation died, one patient with null/null genotype died at 0.19 years. Sixty per cent of patients with other/other and 64% with null/other mutations died at a median age of 0.4 years and 0.13 years, respectively (table 1, see online supplementary table S2, figure 2). Treatment with surfactant was applied to 14 patients, without effect in 8 patients, and with moderate, but transient improvement in 4 patients. Fourteen patients were treated with systemic steroids, showing no effect in eight of the patients, while three patients displayed moderate improvement, and one good improvement. Treatment with hydroxychloroquine was administered to eight patients, in three patients without response, while two patients responded well, and three moderately (see online see online supplementary table S2).
Phenotype groups of patients with ABCA3 mutations
To expand knowledge on various phenotypes of DPLD induced by compound heterozygous ABCA3 mutations, case reports of five patients with prolonged survival are presented in online supplement A.
Phenotype groups
With respect to follow-up we observed two phenotypes: patients with early lethal mutations, either (1a) dying within the first 6 months or (1b) before the age of 5 years, and patients with prolonged survival (2) into childhood, adolescence or adulthood (table 1). All but one patient with null/null mutations were in group 1a, patients with null/other and other/other mutations were distributed over all groups.
Surfactant proteins in BALF
Surfactant analysis in 12 patients with homozygous ABCA3 mutations showed no mature SP-C, in one patient abundant SP-C processing forms were detected. SP-B was always present in variable levels (see online supplementary tables S6 and S7). In patients harbouring compound heterozygous ABCA3 mutations, only two patients had normal SP-C levels in relation to SP-B, all other patients had reduced SP-C levels.
Histology and imaging
Histopathological findings in 14 homozygous patients and 10 compound heterozygous patients revealed patterns typical for non-specific interstitial pneumonia (NSIP), desquamative interstitial pneumonia (DIP), pulmonary alveolar proteinosis (PAP) (alone or in combination) or chronic pneumonitis of infancy (CPI). Notably, all patients with CPI (four homozygous and one compound heterozygous) died under 6 months of age. In one patient an usual interstitial pneumonia (UIP) pattern was seen. Electron microscopy imaging showed electron-dense bodies typical for ABCA3 deficiency. Interestingly, one patient with homozygous ABCA3 mutation had few well shaped lamellar bodies with membranes arranged in parallel. Type II pneumocyte hyperplasia, alveolar macrophage accumulation and alveolar septal thickening were the predominant features in 12 available slides. Furthermore, mild or moderate interstitial chronic inflammation and alveolar septal inflammation were recorded. Interstitial fibrosis was also frequent, however, not present in all cases, appearing rather with increasing age (tables 2 and 3).
Histology and electron microscopy of patients with homozygous ABCA3 mutations
Histology of patients with compound heterozygous ABCA3 mutations
Lung biopsies obtained in 12 of the patients were used for immunohistochemical staining of ABCA3 protein (see online supplementary table S8). Staining intensity and pattern were scored in comparison to control tissues. All controls displayed ring-like structures and huge aggregations of ABCA3 protein. By contrast, staining was weak or absent in patients with ABCA3 mutations. Moreover, aggregates were absent or small and staining pattern was diffuse if present at all. In three patients with null/null mutations, immunohistochemistry was negative for ABCA3, while two patients with other/other mutations had a moderate staining intensity and at least small ABCA3 aggregates (figure 3, see online supplementary table S8 and supplementary figure S1).

{kind=link}
{kind=link}
{kind=link}
Results of immunohistochemical analysis and semiquantitative scoring in 12 patients after staining for ATP-binding cassette subfamily A member 3 (ABCA3). The hypothesis that frequencies of ABCA3 staining pattern, ABCA3 aggregates and the intensity of ABCA3 staining were different between normal subjects and patients carrying ABCA3 mutations was tested using the Fisher's exact test. **p<0.01, ***p<0.001, ***p<0.0001.
CXRs in homozygous and in compound heterozygous patients show predominant ground glass, reticular pattern and bronchial wall thickening (the latter two rather increasing with age). In some cases and to a milder degree, cysts, hyperinflation, nodular pattern and consolidation were noted (see online supplementary tables S9 and S10).
Also in the CTs of homozygous and compound heterozygous patients ground glass opacity and reticular pattern were the predominant features, in some cases a mild degree of cysts, bronchial wall thickening, honeycombing, paraseptal emphysema, crazy paving, nodular pattern or consolidation were noted (see online supplementary table S11).
Peripheral bronchial dilatation, resembling bronchiectasis according to the definition by Hansell et al,28 was noted especially in homozygous patients <0.1 year, possibly due to severe interstitial lung disease and/or severe mechanical ventilation pressure (see online supplementary tables S9, S11 and supplementary figure S2). In one patient alive at 7 years beginning bronchiectasis was noted with progressive interstitial lung disease (see online supplementary figure S3). Due to the limited number of available investigations in sufficient quality, imaging data have to be interpreted with caution.
Discussion
We characterised the spectrum of lung disease caused by ABCA3 mutations in a cohort of 40 patients with two disease-causing mutations. Of these, 19 mutations have not yet been described to our knowledge. We observed two main phenotypes: patients with early lethal mutations, either (1a) dying within the first 6 months or (1b) before the age of 5 years, and patients with prolonged survival (2) into childhood, adolescence or adulthood (table 1).
The most frequent presentation of ABCA3 mutations in our cohort was neonatal RDS in full-term newborns with fatal outcome. Patients presenting in childhood or later9–11 had the typical signs and symptoms of chronic interstitial lung disease. Wambach et al7 recently demonstrated in 185 patients that both, age at presentation and outcome are closely related to the type of mutations (null vs other). The results were confirmed by our data: all patients with null/null mutations presented at birth and died within the first few weeks of life (phenotype group 1a), while some patients with null/other or other/other mutations presented at an older age and survived longer (table 1, figure 2). Thus, while two null mutations seem to be incompatible with life, other mutations can vary significantly in their phenotypical presentation and outcome. However, even identical mutations in siblings are known to vary in phenotype.29 Potential reasons for this variation may be differences in penetrance, epigenetic and environmental factors, and other compensatory mechanisms, possibly within the surfactant system itself.
While PAP is a known histological pattern in ABCA3 mutation, often combined with NSIP or DIP,21 the clinical presentation of neonates is frequently labelled as PAP. This was the case in 11 patients of this cohort. They had either white lungs on CXRs or crazy paving pattern on CT scans with respiratory distress despite being born on time. This clinically dominated labelling should be avoided as none of these patients had a pure PAP pattern in histology. Further, clinical presentation of PAP due to mutations in CSF2RA is beyond the neonatal period.30 Of interest is patient 31 who carried two likely disease-causing variations in the ABCA3 gene who presented with exercise intolerance and some ground glass opacities on CT scan. She developed the typical features of granulocyte macrophage colony-stimulating factor (GM-CSF) autoantibody positive, adult PAP and was in remission after treatments with whole lung lavages (see online supplementary table S2, supplement A).
It is further of interest to note that even in the absence of SP-C in BALF patients may survive into adulthood (eg, patients 11 and 12).10 This suggests that SP-C deficiency may not be the primary disease mechanism in ABCA3 deficiency. On the other hand, the absence of SP-C in BALF may be used as an important indicator for ABCA3 mutations, as has already been suggested by Flamein et al.8
Additional clinical hints for the presence of ABCA3 mutations are given by radiological and histological investigations (tables 2 and 3, see online supplementary tables S9–S11), although they are not very specific. Immunohistochemical staining pattern and intensity correlates with the type of ABCA3 mutation and survival (figure 3, see online supplementary table S8). Absent staining or abnormal staining pattern (diffuse instead of ring-like) is suggestive of ABCA3 deficiency. Sensitivity and specificity need to be investigated in larger numbers of patients and disease controls.
Efficacy of therapeutic interventions in patients with ABCA3 mutations appears to be poor. Our data showed moderate or sometimes good responses to exogenous surfactant in some patients. Treatment with systemic steroids yielded positive effects in 10/32 patients and hydroxychloroquine in 9/15 patients; most effects were transient and overall survival was low. It is important to recognise that prolonged survival can occur in individuals who do not carry the null/null genotype. One recent report also describes significant improvement including delisting for LTX with the application of triple therapy with intravenous pulse methylprednisolone, and enteral hydroxychloroquine and azithromycin,31 suggesting potential efficacy of combination therapy. New therapeutic developments should focus on the underlying mutations and mechanisms. Stop mutations might be addressed with compounds allowing read through,32 whereas missense mutations might be rescued by corrector-type medications.33 The latter may also be addressed by hydroxychloroquine, although its precise mechanism of action is unknown. In vitro studies identifying appropriate candidates are warranted.
Acknowledgments
The authors thank Ms Traudl Wesselak and Andrea Schams for the strong support in collecting and gathering the data. The authors also thank all patients and families for participating in the study, and all members of the ABCA3-study group of the Kids Lung Register for their work and for including patients into the study: P Ahrens, Darmstädter Kinderkliniken Prinzessin Margaret, Darmstadt, Germany; D Aschmann, University Hospital Dresden University, Dresden, Germany; T Berger and N Regamey, Children's Hospital Lucerne, Lucerne, Switzerland; L Chevret, Bicetre Hospital, Le Kremlin Bicetre, France; N Cobanoglu, Ankara University School of Medicine, Ankara University, Ankara, Turkey; JM Cobben, University Hospital, Amsterdam University, Amsterdam, Netherlands; L Donato, University Hospital, Strasbourg University, Strasbourg, France; M Gerstlauer, Klinikum Augsburg, Augsburg, Germany; M Griese, A Irnstetter, M Kappler, M Klemme, C Kröner, F Nagel, M Feilcke, I Pawlita, J Ripper, M Hengst, M Forstner, University Hospital, Munich University, Munich, Germany; K Kristensen, Ringhospitalet, Kopenhagen, Denmark; I Campo, M Luisetti, Clinica di Malattie Apparato Respiratoria, Pavia, Italy; E Mildenberger, University Hospital, Mainz University, Mainz, Germany; G Mostafa and R Weitzdoerfer, University Hospital, Wien University, Wien, Austria; E Eber, A Pfleger, Medical University of Graz, Graz, Austria; L Pinheiro, Hospital Braga, Braga, Portugal; J Pöschl, University Hospital, University Heidelberg, Germany; M Proesmans, UZ Leuven, Leuven, Belgium; J-W Richter, Kinderkrankenhaus auf der Bult, Hannover, Germany; V Rigourd, Institute De Puericulture, Paris, France; K Runge, Klinikum Wuppertal GmbH, Wuppertal, Germany; T Schaible, University Hospital, University Mannheim, Germany; W Schneider, University Hospital, Frankfurt University, Germany; E Cloppenburg, T Hübner, J Seidenberg, Kinderklinik Klinikum Oldenburg, Medizinischer Campus Universität Oldenburg, Germany; T Sismanlar and Ayse T Aslan, Gazi University Hospital, Ankara University, Ankara, Turkey; S C Sloot, Amsterdam Medical Center, Amsterdam, Netherlands; N Schwerk, Medizinische Hochschule Hannover, Hannover University, Hannover, Germany, Y Steinrücke, Klinikum Dritter Orden, München, Germany; S Terheggen-Lagro, Emma children's hospital, Academic Medical Center, Amsterdam, Netherlands, S Wagner; Kinderkrankenhaus St Marien, Landshut, Germany; S Wellmann and C Bührer, University Hospital Basel, Basel University, Switzerland; J Wintgens, Staedtische Kliniken Moenchengladbach, Moenchengladbach, Germany; E Fischer and P Lasch, Klinikum Bremen Mitte, Bremen, Germany; H Teschler, Ruhrland Klinik, Essen, Germany; H Schroten, University Hospital, University Düsseldorf, Düsseldorf, Germany.
References
Footnotes
Contributors MG designed the study, collected the cases, analysed the data and drafted and revised the manuscript. He is the guarantor of the entire manuscript. CK organised and analysed the data and drafted the manuscript. MKl, MH, MKa, NC, TS, ATA, IC, MP, TS, ST-L, NR, EE, JS, NS, CA, CK and MG contributed and evaluated patients and performed chart review. RZ, DR and TW performed laboratory and immunohistochemical analyses, FB and SR performed the histological investigations, VT the radiological investigations. CA and PL performed genetic analysis and contributed to the long-term collection of subjects. All contributors read and critically revised the manuscript and agreed to the final version of the manuscript.
Funding The work of MG was supported by the Deutsche Forschungsgemeinschaft Gr970/8-1, chILD-EU (FP7, No. 305653) and the “Else Kröner-Fresenius-Stiftung” grant 2013_A72.
Competing interests None declared.
Ethics approval Institutional Review Board (EK 026-06), chILD-EU (EK 111-13).
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves