Article Text

Abstract
Introduction Primary ciliary dyskinesia (PCD) is a genetic disease characterised by abnormalities in ciliary function, responsible for chronic pulmonary and sinonasal diseases. Adult clinical features and outcome are poorly described.
Objectives To assess the clinical characteristics and disease progression in adults with PCD.
Methods Bicentric retrospective study, focusing on adults (≥18 years) with an asserted diagnosis of PCD based on the presence of bronchiectasis with typical ultrastructural defect of cilia and/or situs inversus (SI). Clinical symptoms, respiratory function, extent of bronchiectasis, microbiology and molecular analysis were assessed. Results are expressed as median (25th; 75th centile).
Results 78 patients were included with a median follow-up of 8.1 years. 91% of patients had respiratory symptoms and 95% had chronic rhinosinusitis. Half of ultrastructural defects concerned dynein arms. Respiratory function was significantly lower in women (FEV1=60% predicted (50; 76), vs 77% (62; 95), p=0.009) and in patients with chronic airway Pseudomonas aeruginosa (PA, n=21) infection (FEV1=60% (48; 71) vs 75% (55; 89), p=0.04). FEV1 was associated with gender (regression coefficient for men =13.8, p=0.009), chest CT score (r=−0.42, p<0.001) but not with age at diagnosis, SI or body mass index. FEV1 decline was −13.4 mL/year (−42.8; +11.9) and was greater in women (−29.3 mL/year, (−59.7; −11.9), vs –2.0 mL/year (−26.9; +25.4), p=0.002). Three patients had severe respiratory failure.
Conclusions Alteration of respiratory function in adults with PCD is heterogeneous and usually moderate but appears more severe in women and in patients with chronic PA infection. Only 4% of patients develop chronic respiratory failure.
- Bronchiectasis
- Rare lung diseases
Statistics from Altmetric.com
Key messages
What is the key question?
What are the respiratory phenotype and disease progression in adult patients with primary ciliary dyskinesia (PCD)?
What is the bottom line?
In adult patients with PCD, FEV1 is moderately altered and independently associated with female gender.
Why read on?
First description of clinical features including sterility and functional alteration in an adult cohort of PCD.
Introduction
Primary ciliary dyskinesia (PCD) is a rare genetic disorder characterised by abnormal ciliary structure and/or function. PCD is associated with situs inversus (SI) (ie, Kartagener syndrome) in approximately half of the cases.1 The diagnosis of PCD is based on the presence of a suggestive clinical presentation and demonstration of abnormal ciliary motility classically associated with ultrastructural defects of the respiratory cilia. To date, more than 30 genes have been implicated in PCD and identification of mutations in such genes is increasingly used to comfort the diagnosis of PCD.2 Due to the distribution of ciliated cells, the disease involves the whole respiratory tract, usually responsible for a sinopulmonary syndrome. Pulmonary symptoms include chronic cough and sputum with recurrent tracheobronchial infections, often leading to obstructive lung disease and to bronchiectasis. Involvement of the upper respiratory tract is characterised by otitis media with effusion, chronic rhinosinusitis with or without polyps.3
Outcomes of the disease in adulthood is poorly known since, to date, most studies have focused either on children or on both children and adults and thus the decline in respiratory function may be underestimated.4–12 In the same way, fertility status has never been described in a large cohort. Hence, we conducted a retrospective study in two French hospitals in charge of adult patients with PCD for many years. We analysed clinical, radiological and microbiological aspects of the disease, and assessed the longitudinal pulmonary function decline in 78 patients with a definitive and well documented diagnosis of PCD.
Material and methods
Subjects and cilia analysis
We performed a retrospective longitudinal study in 78 adult (≥18 years) patients with PCD recruited in two French tertiary hospitals (Centre Hospitalier Universitaire Cochin, Paris, and Centre Hospitalier Intercommunal, Créteil). Patients were referred to our institutions for chronic and recurrent respiratory symptoms between 1990 and 2010. We considered that a patient had an asserted diagnosis of PCD based on at least one of the following criteria: (1) a clinical diagnosis of Kartagener syndrome (ie, chronic sinusitis, bronchiectasis and SI) and (2) the evidence of PCD-specific ciliary ultrastructural defects or the identification of non-ambiguous causal mutations in a PCD gene in a patient with bronchiectasis on CT scan.
Ciliary investigations were performed once for diagnostic purposes in patients free from respiratory tract infection or exacerbation for at least 6 weeks. Airway ciliated cells were obtained by brushing the middle part of the inferior turbinate or a main bronchus with a 2 mm cytology brush (Laboratoires Gyneas, Goussainville, France). Cells were suspended in B1 bovine serum albumin (BSA) medium (Laboratoire CCD, Paris, France) and examined by microscopy or high speed video microscopy (HSV) for diagnostic procedures after 2010 within 3 hours at room temperature.
All observations were performed within 20 min at 37°C with an inverted microscope (Axiovert 200, Carl Zeiss S.A.S. Le Pecq France), using an oil immersion ×100 objective. Since 1998, beating ciliated edges were recorded with a digital camera (PixeLINK A741, Ottawa, Canada) at a rate of 355 frames per second. Twenty distinct areas containing intact undisrupted ciliated epithelial edges >50 μm, devoid of mucus and beating in the plane of the camera were recorded. Qualitative and quantitative evaluations of ciliary beat pattern were performed as previously described.13 HSV analysis was considered abnormal when the qualitative evaluation showed a beat pattern virtually immotile and/or the measured weighed distance travelled per second was below 24 µm.
Biopsies were obtained from either bronchial (main bronchus) or nasal (inferior turbinate) mucosa and processed for electron microscopy (TEM) as previously described.14 Ciliary ultrastructural results were expressed as a percentage of abnormal cilia among the total number of cilia analysed. For each ciliary ultrastructural study, axonemal abnormalities were quantified, and the ultrastructural phenotype was defined by the main ultrastructural defect (involving the dynein arms or the central microtubules). Dynein arms were considered to be absent from axonemal sections when the structure was missing from at least five of the nine peripheral doublets. The central structures (central microtubules and central sheath) were termed «central complex». As previously reported,14 up to 10% of cilia in control specimens can exhibit ultrastructural defects. For this study, ciliary abnormalities were defined as the presence of at least 20% of ciliary defects.
Since 2004, nasal NO (NOn) was measured according to international guidelines,15 using a chemiluminescent nitric oxide analyser (EVA4000, Series, Aix en Provence, France), as previously described. Results were expressed as NOn output (nL/min) and NOn <200 nL/min was considered to be abnormally low, as frequently reported in PCD.8 ,10 ,11
Genomic DNA was obtained from whole blood samples by use of a FlexiGene kit (Qiagen, France). The molecular analysis was stopped when two unambiguous molecular defects were identified in the homozygous or compound heterozygous state.
Data collection
All data were obtained during the usual follow-up of patients. No supplemental examination was performed for this study.
Calculation of annual decline in FEV1
For each patient, all FEV1 values were recorded after the age of 18 years to acknowledge for adulthood respiratory function, according to European Respiratory Society/American Thoracic Society (ERS/ATS) recommendations.16 Postbronchodilator FEV1 was used and FEV1 decline was calculated if there were three or more values of FEV1 and a follow-up of at least 2 years. According to the European Coal and Steel Community (ECSC)/ERS 93′ reference equation,17 we used a linear equation to calculate the annual decline in FEV1 for each patient.
Microbiological findings
All sputum cultures available were used. Each patient has at least a sputum bacteriological sample every 3 months. A patient was considered chronically infected with a bacteria when at least three sputa samples in <6 months were positive.18
CT scoring system
Chest and sinus CT were performed on a routine basis. For chest CT scoring, we used the modified Bhalla score for bronchiectasis.19 For each patient, the most recent chest CT was used to assess the score. In patients with SI, we considered the lung in which the middle lobar bronchus and the corresponding middle lobe were identified on the CT scan as the right lung. The scores for the six lobes were then summed to provide a total CT scan score, with a theoretical range from 0 (normal) to 48 (maximal score in all lobes).
For sinonasal disease, we used the Lund-MacKay staging system,20 on the last available CT. We assessed a score ranging from 0 (normal) to 2 (complete opacity) for each of the sinus and the ostiomeatal complex. Total score was calculated by summing the score for each sinus and the ostiomeatal complex with a theoretical range from 0 (normal) to 24 (maximal score).
Statistical analysis
Results were expressed as medians (25th; 75th centiles) or percentages when appropriate. Comparisons between two groups for quantitative data used Wilcoxon test. Normality of the distribution of variables was assessed with the D'Agostino-Pearson test. Pearson's correlation coefficient and Spearman's rank correlation coefficient assessed correlations among variables. p values of <0.05 were considered statistically significant. Stepwise multiple regressions were performed to analyse the relationship between last available FEV1, and body mass index, sex, SI, age, chest-HRCT score. We also performed an analysis of covariance using gender as a factor, body mass index (BMI) and age as cofactors. All analyses were performed using Matlab R2013a software (Mathworks), PRISM 6 software (GraphPad Software) or MedCalc for Windows, V.15.1 (MedCalc Software, Ostend, Belgium).
Results
Characteristics of the PCD population
Characteristics of patients are summarised in table 1. Patient's ages ranged from 18 years to 77 years. Median follow-up at the time of data collection was 8.1 years (3.7; 12.6). Only 17 patients (22%) had a familial history if PCD, among which 6 (7.7%) were born from consanguineous union. Another 11 patients (14%) had only a consanguinity history. Median age at diagnosis of PCD was significantly lower in patients with SI than in those without SI (median age 6 years vs 16 years, p=0.0075). Most patients (69%) had onset of respiratory symptoms in preschool but only 18 (23%) had a definite history of neonatal respiratory distress syndrome. Nevertheless, level of dyspnoea in adulthood often remained mild with 78% of patients classified 0 or 1 on the modified Medical Research Council (mMRC) scale. Of the three patients with retinitis pigmentosa, two were brothers and one had a sibling with PCD but no retinitis pigmentosa at the time of study.
Characteristics of patients
Ultramicroscopic features were available in 62 patients (79.5%) and are presented in table 2. Most patients had a dynein arm defect, mostly outer dynein or both dynein arms. The other 16 patients had SI with impossible ultrastructural analysis (no or altered cilia in samples).
Ultramicroscopic features of patients with primary ciliary dyskinesia
Molecular analyses were performed in 60 patients (77%) and non-ambiguous mutations were identified in 40 patients (table 3); in all patients the implicated gene was consistent with the ultrastructural phenotype. Only two patients had Kartagener syndrome and no ultrastructural defect nor mutation.
Genetic findings in patients with primary ciliary dyskinesia
NOn level was available in 40 patients (51%). Median NOn level was 18.9 nL/min (11.0; 95.7). Three patients (7.5%) had a NOn >200 nL/min: two with outer dynein arm defect and one with Kartagener syndrome and no ultrastructural defect evidenced by TEM.
To summarise diagnostic results, among the 37 patients with Kartagener syndrome, 20 had PCD-specific ciliary ultrastructural defect and non-ambiguous causal mutation, 8 had PCD-specific ciliary ultrastructural defects without genetic mutation, and 9 had normal ciliary ultrastructure and no genetic mutation. Among the 41 patients without Kartagener syndrome, 20 had PCD-specific ciliary ultrastructural defects and non-ambiguous causal mutation, 19 had PCD-specific ciliary ultrastructural defects without genetic mutation, 2 a normal ciliary ultrastructure without genetic mutation with first-degree relatives with Kartagener syndrome.
Sputum bacteria
Table 4 summarises the bacteria identified at least once in a patient's sputum and the chronic bacterial sputum infection.
Bacteria identified in patients' sputum
Twenty-one patients (27%) had chronic Pseudomonas aeruginosa (PA) infection. Median age at first identification of PA was 32.5 years (18; 44). Median age at PA chronic infection was 39 years (21.6; 49.2). Both were not significantly different in men and women; median time between first identification of PA and chronic infection was 1.2 years (0.5; 2) but was more than 4 years in four patients who had had a first successful eradication of PA in sputum.
Respiratory function of adults with PCD
Respiratory function at the end of follow-up is summarised in table 5.
Respiratory function at the end of the study (n=78)
Two patients had a FEV1<30% (2.5%), 12 patients had 30%≤FEV1<50% (15.5%), 37 patients had 50%≤FEV1<80% (46.1%) and 27 patients had a FEV1≥80% (35.9%).
Respiratory function at the end of the study was significantly lower in women than in men and in patients with chronic PA infection (figure 1A). Bivariate analysis showed that last available FEV1 (% predicted) correlated with body mass index (r=0.2831, p=0.0120, see online supplementary figure S1C), age at the time of study (r=−0.2463, p=0.03, see online supplementary figure S1B) but not with age at diagnosis (r=−0.02, p=0.9), presence of SI (p=0.6) or with ultramicroscopic (dynein arm defects vs central complex defects, p=0.9) or genetic abnormalities (dynein arm defects vs central complex defects, p=0.9). Using stepwise multiple regression with last available FEV1, we found an independent relationship between gender (regression coefficient for men=13.8, p=0.009), chest CT score (regression coefficient=−0.93, p<0.001) and last available FEV1. The difference between genders remained after adjusting for age and body mass index.
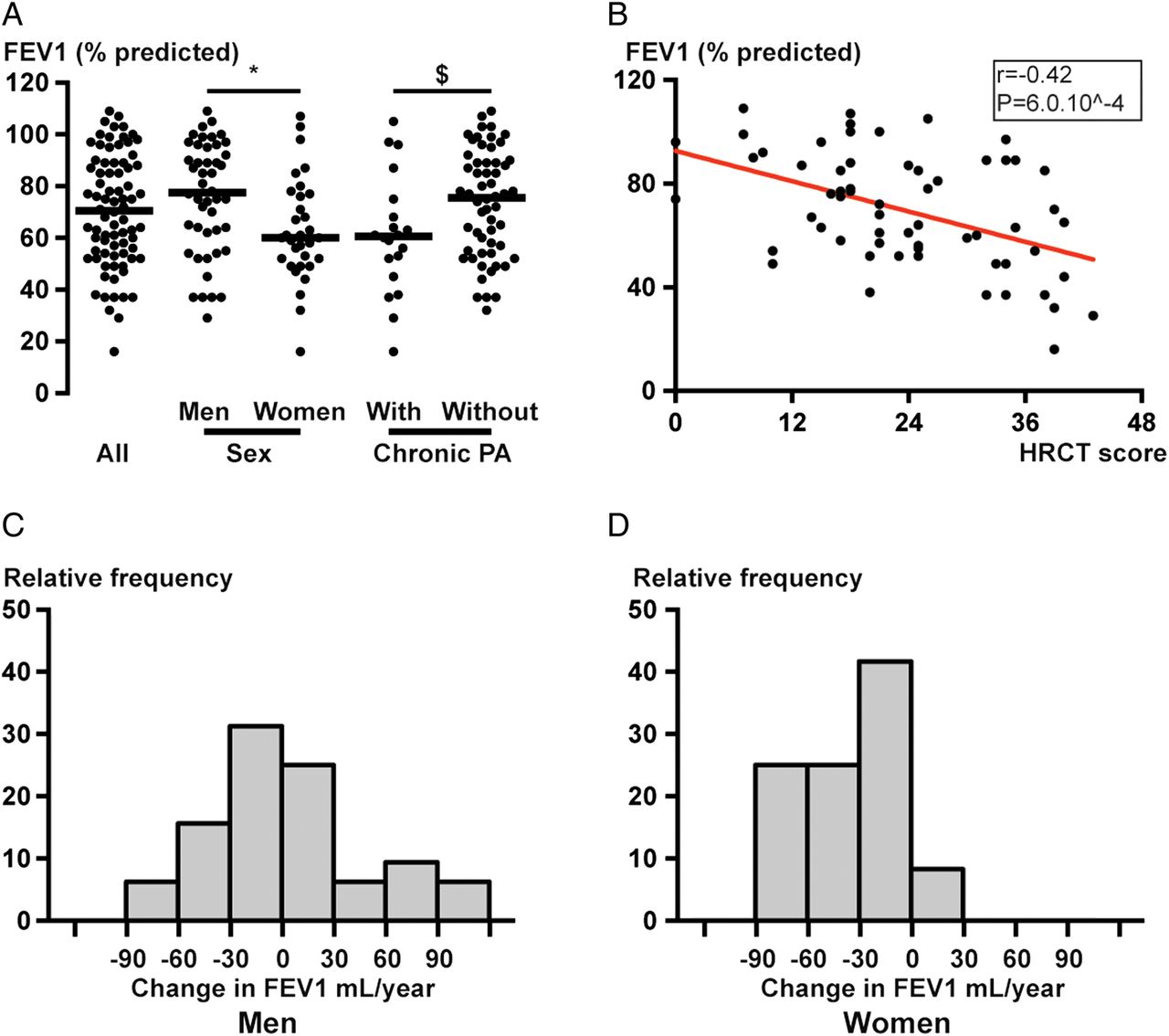
{kind=link}
Respiratory function of patients with primary ciliary dyskinesia (PCD). (A) Shows last available FEV1 (% predicted) for each patient in all patients (left), men, women (middle) and patients with and without chronic Pseudomonas aeruginosa (PA) infection (right). *p=0.009 between men and women; §p=0.04 between patients with and without chronic PA infection. (B) Shows the relationship between FEV1 (% predicted) and total HRCT score on the last HRCT available; Spearman's rank correlation coefficient r=−0.42 and p=6.10−4. (C and D) show the annual evolution of FEV1 (mL per year) of follow-up for men (C) and women (D), calculated using a linear equation for each patient after the age of 18 years.
Supplemental material
Annual decline in FEV1 (mL/year) is presented in figure 1C, D and in online supplementary figure S1A and was significantly greater in women (−29.3 mL/year, (−59.7; −11.9)) than in men (−2.0 mL/year, (−26.9; +25.4), p=0.002). No significant difference in FEV1 decline was observed between patients with and without chronic PA infection (−22 mL/year vs −9 mL/year, p=0.14). FEV1 decline in patients with chronic PA infection was not accelerated after infection (−21 mL/year before vs −24 mL/year after, p=0.81), but only 12 patients could be included in the calculation.
Radiological findings
Sixty-four high resolution computed tomographies (HRCTs) were available for CT scoring in 64 patients. Median total CT score was 22/48 (17; 32.5). There were significantly more radiological lesions in lower lobes, as shown in online supplementary table S1. FEV1 values (% predicted) at the end of study were correlated to chest CT score (r=−0.42, p=6.0 10−4, figure 1B).
ENT findings
Sinonasal clinical assessment was available in all patients, with 74 (94.8%) having confirmed chronic rhinosinusitis and 44 (56.4%) having nasal polyposis. Forty-one (52.6%) patients had had sinus surgery at least once. CT scoring was available in 50 patients, with a median CT score 15/24 (10.8; 19).
Of the patients, 55.1% had a history of otitis media with effusion in their childhood; most patients had normal eardrum examination in adulthood but 16.7% had had tympanostomy tubes at least once and 28.2% exhibited chronic otitis with or without otorrhoea. Only three patients had a history of ear surgery for chronic otitis. History of acute otitis media was less common (40.0% of patients) and again mostly present during childhood.
Treatments
Intravenous antibiotics were given mostly for PA or Stenotrophomonas maltophilia infection, and intravenous courses usually lasted 14 days. Median number of intravenous antibiotic treatment per patient and per year was one. Oral antibiotic prophylactic therapy was used in 24% of patients and was started in childhood in all patients but two, and could be stopped in adulthood in 63% of patients, but was often replaced by long-term macrolides and/or inhaled antibiotics.
Inhaled antibiotics were given in 21 (26.9%) patients, with 11 patients receiving colimycin, 7 patients receiving tobramycin and 3 patients receiving both. Long-term macrolides were given in 25 patients (32%).
Fertility
Thirty-nine patients had never tried to have children at the end of follow-up. Among the remaining 39 patients, presumably 10 men (4 with dynein arm defect, 3 with abnormal central complexes, 2 with multiple defects, 1 with unknown defect but SI) and 10 women (5 with outer or inner dynein arms defects, 4 with unknown defect but SI) had children (25.6% of the cohort) without assisted reproductive technologies.
Fourteen men had hypofertility (10 with dynein arms defect, 3 with abnormal central complexes and 1 with unknown defect) and had seven children after intracytoplasmic sperm injection and seven children after in vitro fertilisation. Five women had hypofertility (four with dynein arms defect and one with unknown defect). We didn't report any ectopic pregnancy but three patients had had miscarriages.
Disease severity
Long-term oxygen was prescribed in four patients and non-invasive ventilation in three patients; only one patient had undergone lung transplant, and another one was on transplant waiting list. Of note, 21 patients had had surgery for bronchiectasis in childhood. Of the two patients with a history of severe haemoptysis, one had required embolisation and the other one lung transplant for recurrent haemoptysis (see above).
Two patients were dead at the time of study, one at 37 years after lung transplant for recurrent haemoptysis (patient died from early bronchial dehiscence) and one who had refused lung transplant and died at 35 years of end-stage respiratory failure.
Discussion
To our knowledge, this is the first study on PCD that focuses on adult patients allowing for a better assessment of the evolution of the respiratory disease throughout life. Our main result is that although median FEV1 annual decline is small, the disease appears more severe in women and in patients chronically infected with PA and can lead to chronic respiratory failure in some patients. FEV1 also correlated with total CT score.
PCD diagnosis remains a challenge as no clinical and paraclinical criteria are specific of the disease. Neonatal respiratory distress was frequent in our patients, although less than in other studies,4 but did not always lead to PCD diagnosis. This is likely due to a lack of specificity of this symptom and the difficulty to obtain this information, based on a recall issue for adults. We show that most patients have preschool onset of symptoms but that diagnosis is often delayed in patients even in the presence of SI. Patients from our study presented mostly with dynein arms defect, confirming previous studies.4 ,6 ,7 ,14 Level of NOn was usually under 100 nL/min but three patients had a normal NOn with several measurements showing that PCD should not be definitively excluded based on a normal NOn level if symptoms are very suggestive of the disease. Of note, unlike in other studies, we did not find a higher NOn level to be associated with central complex defects.7
Pulmonary functional decline in PCD is heterogeneous and we lack consistent clinical or morphological markers to adjust and/or reinforce therapeutics. To date, there is still debate about correlations between genotype and/or microscopy and phenotype in PCD, some authors finding different clinical subtypes while others failed to identify such subtypes in their cohorts.7 ,11 ,21–23 Indeed, Vallet et al7 found that patients with central complex defects had a later onset of symptoms but a more severe respiratory disease, and Knowles et al21 found a less severe disease in patients carrying a RSPH1 mutation, Davis et al23 found lower respiratory function, body mass index and greater radiological severity in patients with central complex or inner dynein arm defects compared with patients carrying outer dynein arm defects. In contrast, no difference was observed by Ellerman et al11 and Boon et al.22 We did not find a relationship between respiratory function and ultrastructural phenotype and/or genotype in our cohort of patients but the small number of patients, as in preceding studies, does not allow speculating about a potential correlation. Our study confirms some of the radiological findings of Kennedy et al5 showing a greater involvement of lower lobes and lingula, but not middle lobe, and, although we did not use the same scoring system, a strong correlation between total CT score and FEV1. Respiratory infections play an important part in PCD symptoms and could be involved in lung function decline. Sputum monitoring is now recommended in children,3 and routinely controlled in our centres. Microbiology of sputum in our study showed that the two most common bacteria were Streptococcus pneumoniae and Haemophilus influenzae, consistent with other studies. Of note, no patient had Burkholderia cepacia or methicillin-resistant Staphylococcus aureus in sputum. Also, unlike Noone et al,4 we found non-tuberculous mycobacteria to be rare and only one of our patients had chronic mycobacterial infection. However, we did not record all negative samplings and this low mycobacterium detection should be confirmed in a prospective study with a standardised frequency of sputum analysis for mycobacteria in all patients.
We found a lower proportion of chronically PA-infected patients in our cohort than in other studies.12 ,24 ,25 The relatively short time between first identification of PA and chronic infection in sputum, especially in older patients, may be influenced by late detection of the first Pseudomonas isolate. It addresses the question of the need for a more aggressive treatment of first PA to obtain eradication, as in cystic fibrosis (CF) but at this time we lack data to justify this decision.
Regarding therapeutic issues, continuous antibiotic therapy was frequent in childhood and could be stopped in adulthood in 63% of patients, however they often are replaced by long-term macrolides and/or inhaled antibiotics.
Up to now, no study has described fertility rate or fertility issue in patients with PCD. Regarding the 39 patients with fertility results in our study, it is of course difficult to speculate about the real fertility rate in men but we confirmed that 10 of the 15 women conceived spontaneously and we did not observe ectopic pregnancy, which has been describe previously as a potential complication of PCD.26 ,27
Annual decline of FEV1 was low (−13.4 mL/years) in our cohort of patients, still 18% of patients had FEV1 of <50% predicted. This annual decline is lower than what has been observed in a non-CF bronchiectasis adult cohort retrospective study (−54 mL/years).28
Women had a significantly lower respiratory function at the end of the study and a steeper decline in FEV1. FEV1 was correlated with age but age was not significantly different between men and women and could not account for this difference, as well as BMI and smoking habit. This gender difference is comparable to the observations made in cystic fibrosis in which the disease is more severe in women.29 ,30 but to our knowledge, had never been described in PCD. FEV1 was significantly lower in patients with chronic PA infection, but unlike in cystic fibrosis,29 the rate of decline in FEV1 was not accelerated in our patients once they had Pseudomonas colonisation. This result should be taken cautiously considering the small number of patients (only 12) that could be included in the analysis and it is possible that a greater length of follow-up in these patients will uncover a greater decline in FEV1.
Limitations of the study were the post hoc collection of data and the change of standard care notably the antibiotic therapy, during the period of the study. It may bias our results with a poorer outcome in patients diagnosed earlier in the study. Moreover, the focus of the study (ie, adulthood) is one of the originality of the study but may ‘exclude’ young patients with PCD who succumbed to disease. However, it has been shown that mortality is very low in PCD and the course of disease in children after diagnosis is independent of age at diagnosis.9
In conclusion our data show a moderate alteration of lung function but 18% of patients had a FEV1 below 50%. Female gender and chronic infection with PA are associated with worse respiratory function. Sterility information was available in 50% of patients and we confirmed that women with DCP can conceive spontaneously.
Acknowledgments
The authors thank Jean-Baptiste Masson for helpful discussion and also the patients.
References
Footnotes
Contributors JF-M and BM had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. BM, JF-M, IH and LB designed the research. JF-M, LB, IH, ND, AC, JFP and EE contributed to the acquisition of data. BM, JF-M, LB, IH and BH analysed the results and made the figures. BM, JF-M, LB, IH, BH, AC, JFP, EE and P-RB wrote the paper.
Competing interests None declared.
Ethics approval Institutional Review Board of the Société de Pneumologie de Langue Francaise.
Provenance and peer review Not commissioned; externally peer reviewed.