Article Text

Abstract
Background Neutrophil extracellular traps (NETs) represent a distinct strategy by which neutrophils trap, confine and eliminate invading microorganisms. Emerging evidence suggests that NETs exert a deleterious effect to the host in the absence of microbial stimuli. However, the role of NETs in smoking-related lung diseases remains to be elucidated.
Objectives To evaluate the formation of NETs in the context of chronic inflammation induced by cigarette smoking and explore its potential role in an experimental mouse model of emphysema.
Methods The formation and degradation of NETs in cigarette smoke exposed mice was assessed with a fluorescence microscope. The potential influences of NETs on plasmacytoiddendritic cells were also investigated.
Results NETs were more prone to formation by polymorphonuclearneutrophils but defective in degradation in cigarette smoke exposed mice. Cigarette smoke extract (CSE) served as an important facilitator that triggered neutrophils to undergo NETosis in vitro. Furthermore, CSE-induced NETs were capable of driving plasmacytoiddendritic cell maturation and activation, thereby initiating a T-cell-mediated immune response.
Conclusions NETs may represent a critical connection between innate and adaptive immune responses under conditions of chronic inflammation induced by cigarette smoke exposure.
- neutrophil extracellular traps
- plasmacytoid dendritic cells
- cigarette smoke
Statistics from Altmetric.com
Key messages
What is the key question?
Do neutrophil extracellular traps (NETs) play a role in chronic inflammation induced by cigarette smoke exposure?
What is the bottom line?
NETs are abundantly produced by activated polymorphonuclear neutrophils from mice exposed to cigarette smoke, which may cause tissue injury, and more importantly , initiate a T-helper-cell-mediated immune response.
Why read on?
NETs may serve as a facilitator that contributes to chronic inflammation induced by cigarette smoke exposure.
Introduction
Cigarette smoking-related lung diseases (including chronic bronchitis, chronic obstructive pulmonary disease (COPD) and emphysema) are characterised by chronic neutrophilic airway infiltration. The early event that triggers airway inflammation is the chronic activation of the innate immune system in response to cigarette smoke. Neutrophils are the first line of innate host defense against invading bacteria and pathogens. Formation of neutrophil extracellular traps (NETs) has been recently proposed to represent a distinct strategy by which neutrophils capture bacteria extracellularly and kill invading microorganisms.1 2 NETs are a meshwork of DNA fibres released by activated polymorphonuclear neutrophils (PMNs) and decorated with antimicrobial proteins, and enzymes such as neutrophil elastase and myeloperoxidase from neutrophil granules.1 Although NETs function as an effective antimicrobial barrier, excessive formation of NETs could act as a double-edged sword by contributing to organ damage.3 4 In addition, extensive NETs formation has been implicated in the pathogenesis of many autoimmune diseases.3 4 Recent reports reveal that NETs were abundantly generated in the sputum of patients with stable COPD and correlated with the severity of airflow limitation, which suggests NETs play and important role in chronic airway inflammation and lung damage due to COPD.5 6 However, the formation and function of NETs in the context of chronic inflammation which is typically in smoking-related lung diseases remain largely unknown.
The transient and non-specific property of innate immunity fails to fully account for the development of COPD, which progresses even long after smoking cessation, suggesting an adaptive immune component might drive the chronic and ongoing inflammation of this disease. Indeed, recent studies provided direct evidence that adaptive immune response has a crucial role in the persistence of inflammation in COPD.7 Our studies and previous observations have demonstrated that T cells in patients with COPD and in mice exposed to cigarette smoke exhibited predominantly T helper 1 (Th1)/T helper 17 (Th17) patterns, with increased expression of interferon (IFN)-γ and interleukin (IL)-17 that correlated with the degree of airflow obstruction.8–10 Even though antigen-specific T-cell subsets play a critical role in COPD, the upstream molecular events that trigger these immune responses are elusive.
Plasmacytoid dendritic cells (pDCs) are a small subset of dendritic cells (DCs) bridging the innate and adaptive immune responses. Neutrophils were recently identified as a key cell in the orchestration of adaptive immunity by modulating DC functions.11 Actually, previous studies have shown that the immunogenic complexes composed of LL-37 and self-DNA produced by activated neutrophils efficiently trigger pDC activation in patients with psoriasis and systemic lupus erythematosus (SLE).12 13 Interestingly, several investigators have reported that conventional pulmonary DCs and pDCs accumulate in airways and lung parenchyma and exhibit a maturation phenotype in patients with COPD.14–16 Moreover, lung myeloid dendritic cells derived from smokers can direct the generation of pathogenic T cells.17 Nevertheless, the mechanisms by which cigarette smoke switch normal lung DCs to an aggressive form in smoking-related COPD have not yet been fully elucidated.
In this study, we have shown that cigarette smoke exposure elicited exaggerated Th1 and Th17 responses accompanied by chronic activation of pDCs in mice. Additionally, NETs were abundantly released by activated polymorphonuclear neutrophils from mice after chronic cigarette smoke exposure, whereas they were defective in degradation by serum from cigarette smoke exposed mice. In vitro, cigarette smoke extract (CSE) directly induced the formation of NETs. Finally, NETs induced by CSE were found to effectively promote pDC activation, thus introducing the generation of Th1 and Th17 responses by naive CD4+ T cells. These findings identified a critical connection between innate and adaptive immunity in which NETs induced by cigarette smoke potentially activated pDCs and then initiated a specific T-cell-mediated immune response, thus providing insight into the pathogenesis of smoking-related COPD and important therapeutic targets.
Materials and methods
Details are provided in the online Supplementary 1.
Animals and CSE
Male BALB/c mice were purchased from the Guangxi Medical University Laboratory Animal Centre (Nanning, China) and were exposed to cigarette smoke as we previously described.18
Evaluation of experimental mouse model of emphysema
Mice were anaesthetised and sacrificed 24 hours after the last cigarette smoke exposure. The enlargement of alveolar spaces was assessed by determining the mean linear intercept (Lm).
Mouse immune cell preparation and isolation
Mouse blood samples were collected in EDTA-treated tubes via retro-orbital bleeding. Peripheral blood mononuclear cells (PBMCs) and PMNs were isolated by mouse peripheral blood neutrophil separation medium (TBD; China). pDCs and naive CD4+ T cells from spleen single-cell suspensions were purified by magnetic negative bead selection according to the manufacturer (Miltenyi Biotec, Aubum, California, USA). Mice lung single-cell suspensions were isolated by modifying established protocols, using a combination of mechanical fragmentation, enzyme digestion, and centrifugation procedures as described previously.8 10 18
Flow cytometry
Surface molecule and intracellular cytokine detection was performed by flow cytometry. For more details, see online supplementary file.
Preparation of CSE
CSE was prepared by using a modification of the method as we previously described.18
Induction of NET formation and immunofluorescence
NETs were induced as previously described.1 13 Briefly, freshly isolated PMNs were seeded on poly-D-lysine coated coverslips in 24-well round bottom culture plates (1.0×106 cells) in 500 µl serum-free RPMI 1640, and allowed to settle down for 1 hour. PMNs were stimulated with PMA (100 nmol/L) or 0.3% CSE for 4 hours in a CO2 incubator at 37°C. Cells were fixed in 4% paraformaldehyde and then permeabilised with 0.5%Triton X-100. To visualise NETs, the DNA was stained with propidium iodide (PI) and examined with fluorescence microscope.
Preparation of NETs and stimulation of pDCs
NETs were prepared as previously described.19 dsDNA in NETs was quantified by Picogreen dsDNA Assay kit (Invitrogen; P7589) according to the manufacturer’s instructions.
To determine the effect of CSE-induced NETs on pDC maturation, primary pDCs isolated from spleens of air-control mice or pDCs isolated from cigarette smoke exposed mice were plated in 24-well plates at a density of 1×106 cells/mL in the presence or absence of CSE-induced NETs for 18 hours. Subsequently, pDC surface molecules were determined by flow cytometry, and supernatant of culture mediums was used for cytokine measurement. In some experiments, pDCs were collected for assessing the capacity to promote the differentiation of naïve CD4 T lymphocytes.
In vitro pDCs stimulated with CSE-induced NETs were cocultured with naive CD4 + T lymphocytes isolated from spleens of air-control mice (1:1 ratio) in 24-well plates. Four days later, cells were stimulated with 25 ng/mL PMA and 1 ug/mL ionomycin in the presence of GolgiStop for 4 hours. Cells were then collected and stained for surface markers PerCP-CD4 followed by intracellular labelling of PE-IL-17 and APC-IFN-γ. Alternatively, some cells were permeabilised with Foxp3/Transcription Factor Staining Buffer Set and intracellularly stained with PE-anti-mouse T-bet and APC-anti-mouse ROR-γt.
Statistical analysis
Data were expressed as medians. Differences between groups were compared using a non-parametric test (Mann-Whitney test between two groups or Kruskal-Wallis one-way ANOVA on ranks between different groups). Analysis was completed with SPSS version 16.0 Statistical Software, and p<0.05 was considered statistically significant.
Results
Th1 and Th17 responses were exaggerated in mice after chronic cigarette smoke exposure
Using an experimental mouse model of emphysema, we observed that 6 months of cigarette smoke exposure dramatically increased the volume of alveolar spaces accompanied by the destruction of alveolar walls, which typically occurs in patients with COPD (figure 1A). The median of linear intercept (Lm) values in cigarette smoke exposed mice was significantly higher than in air-control mice (46.6 vs 31.5 µm, respectively; p<0.01) (figure 1B). Flow cytometry was performed on CD3+ CD4+ T cells after intracellular cytokine staining with IFN-γ and IL-17 (figure 1C). The representative flow cytometric dot plots of Th1 and Th17 cells are shown in figure 1D. We did not find statistical differences in the overall abundance of CD3+ CD4+ T cells in peripheral blood, spleens and lungs between air-control and cigarette smoke exposed mice (figure 1E). However, the frequencies of Th1 and Th17 cells have shown a significant increase in cigarette smoke exposed mice compared with air-control mice (figure 1FG). These results collectively demonstrated that Th1 and Th17 responses are both exaggerated in an experimental mouse model of emphysema induced by chronic cigarette smoke exposure.

Chronic cigarette smoke exposure induced exaggerated T helper (Th)-1 and Th17 responses in mice. (A) Chronic cigarette smoke exposure caused an emphysematous phenotype by showing enlargement of the alveolar spaces and destruction of the alveolar walls (magnification, ×200). The qualitative assessment of emphysema was determined by measuring the linear intercept (Lm). (B) Comparison of Lm values in air-control and cigarette smoke exposed mice, n=8 mice/group. (C) Lymphocytes were identified based on their characteristic properties shown in the FSC and SSC, and CD3+ CD4+ T cells were gated on lymphocytes. FSC, forward-scattered light; SSC, side-scattered light. (D) The representative flow cytometric dot plots of IFN-γ+ CD4+ Th1 cells and IL-17+ CD4+ Th17 cells in peripheral blood, spleen and lung of air-control and cigarette smoke exposed mice. (E) Comparisons of proportions of CD4+ T cells in peripheral blood, spleen and lung of air-control and cigarette smoke exposed mice. (F) Comparisons of percentages of Th1 cells and Th17 cells (G) Th17 cells in peripheral blood, spleen and lung of air-control and cigarette smoke exposed mice. Data are expressed as medians. The comparisons were determined by Mann-Whitney test. **p<0.01. NS, not significant.
Chronic cigarette smoke exposure was associated with sustained activation of pDCs in mice. pDCs were isolated from spleens of air-control or cigarette smoke exposed mice by negative selection with magnetic microbeads. The maturation of pDCs was determined by assessing the expression of MHC class II and costimulatory molecules CD40 and CD86. The representative flow cytometric histograms of CD40, CD86 and MHC-Ⅱ on pDCs in air-control or cigarette smoke exposed mice are presented in figure 2B–D. The levels of costimulatory molecules CD40 and CD86 on pDCs were significantly increased in cigarette smoke exposed mice compared with air-control mice (figure 2E–F). Moreover, the expression of MHC− IIon pDCs isolated from cigarette smoke exposed mice was significantly higher than air-control mice(figure 2G). Together, these observations suggest that pDCs in chronic cigarette smoke exposed mice are exhibiting a sustained activation phenotype.

Cigarette smoke exposure was associated with chronic activation of plasmacytoid dendritic cells (pDCs) in mice. pDCs were isolated from spleens of air-control or cigarette smoke exposed mice by negative selection with a Plasmacytoid Dendritic Cell Isolation Kit (Miltenyi Biotec). (A) The purity of mPDCA-1+ pDCs was >90%. The levels of MHC class II and costimulatory molecules CD40 and CD86 were determined by flow cytometry. The representative flow cytometric histogram of (B) CD40, (C) CD86 and (D) MHC-II on pDCs in air-control or cigarette smoke exposed mice (n=5 mice/group). Comparisons of frequencies of (E) CD40+, (F) CD86+and (G) MHC-II+pDCs in air-control or cigarette smoke exposed mice. Data are expressed as medians. The comparisons were determined by Mann-Whitney test. **p<0.01.
NETs were more prone to formation by PMNs but defective in degradation in cigarette smoke exposed mice
While exploring the mechanisms of NETosis, we observed an interesting phenomenon that neutrophils isolated from cigarette smoke exposed mice were more sensitive to forming NETs in the stimulation of PMA, a non-physiological agonist commonly employed to induce NETosis in vitro. Only few PMNs underwent NETosis without stimulation whether or not they were isolated from cigarette smoke exposed mice (figure 3A–B). However, compared with neutrophils isolated from air-control mice, neutrophils derived from cigarette smoke exposed mice were more capable of forming NETs in response to PMA (figure 3C–E). It seems that neutrophils were equipped with an enhanced function of NET deployment in the setting of chronic cigarette smoke exposure, a consequence that may contribute to excessive, injurious inflammation, which was observed in smoking-related emphysema.
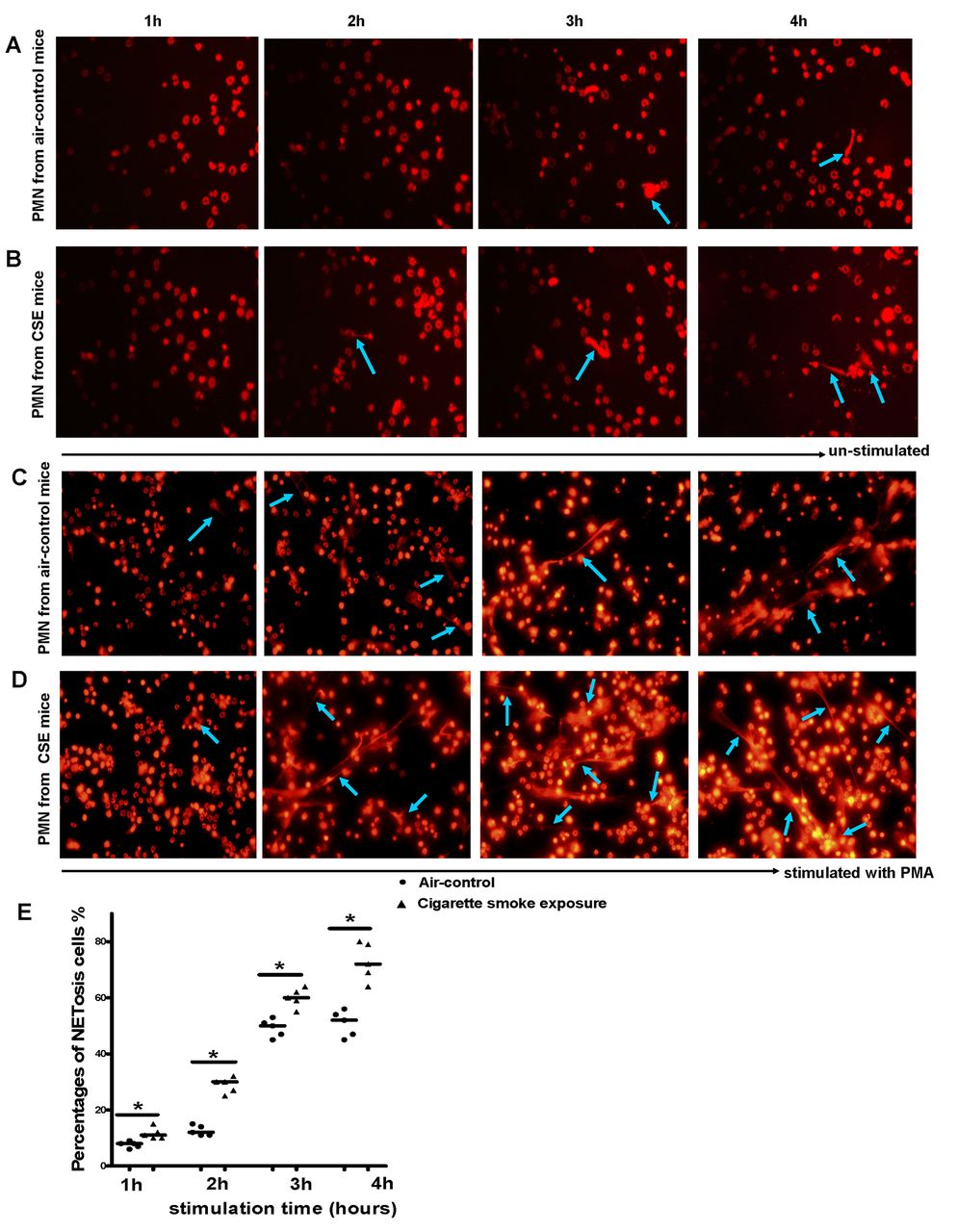
Polymorphonuclear neutrophils (PMNs) isolated from chronic cigarette smoke exposed mice were more prone to formation of neutrophil extracellular traps (NETs). Neutrophils isolated from peripheral blood of (A) air-control or (B) cigarette smoke exposed mice were incubated with medium alone and allowed to spontaneously release NETs at 37°C for 1 hour, 2 hours, 3 hours and 4 hours on glass coverslips coated with poly-D-lysine. Neutrophils isolated from peripheral blood of (C) air-control or (D) cigarette smoke exposed mice were stimulated for NET formation with PMA (100 nmol/L) at 37°C for 1 hour, 2 hours, 3 hours and 4 hours. Neutrophils were then fixed with 4% paraformaldehyde for 20 min at room temperature and stained with propidium iodide (PI). NETosis was then examined by fluorescence microscopy (NETs: blue arrows); the images were representative of five separate experiments. Original magnification, ×40; To quantify NET formation at the microscopical level, 200 neutrophils were analysed in each sample by two independent investigators. In fluorescence microscopy, only cells that feature the morphological characteristics of NETosis (nucleus became flat and associated with chromatin decondensation) were identified as NETosis cells. (E) Comparisons of percentages of NETosis cells in response to PMA between two groups at each time point. Data are expressed as medians. The comparisons were determined by Mann-Whitney test. *p<0.05.
NET-mediated inflammation is strictly confined to local sites, due to the timely degradation of NETs by endonucleases, such as DNase I, which is extremely vital for host homeostasis avoiding unnecessary tissue injury and preventing inappropriate exposure of self-antigens. Thus, the dysregulation of these nucleases might impair the clearance of NETs. To evaluate the NET-degrading activity of serum from cigarette smoke exposed mice, NETs were induced with PMA and exposed to 10% serum from air-control or cigarette smoke exposed mice for 30 min. NET degradation was confirmed by fluorescence microscopy. Strikingly, we found that serum from cigarette smoke exposed mice had an impaired function for degrading NETs; this effect was independent of stimulus (data not shown), whereas treatment with DNAse I induced a rapid degradation of NETs (see online supplementary figure S1).
CSE directly triggered the formation of NETs in vitro
The lack of signs of infection led us to hypothesise whether the susceptibility of PMNs in cigarette smoke exposed mice to form NETs was the direct response to cigarette smoke. To test this possibility, we isolated neutrophils from peripheral blood of air-control or cigarette smoke exposed mice and stimulated them with 0.3% CSE for 4 hours in vitro. As expected, CSE was capable of triggering NET formation which shared similar morphological changes with PMA-induced NETs (figure 4A–B). However, we failed to identify a specific component in CSE accountable for the formation of NETs, since the downstream events (eg, the production of reactive oxygen species (ROS)) induced by CSE could also facilitate NETosis.
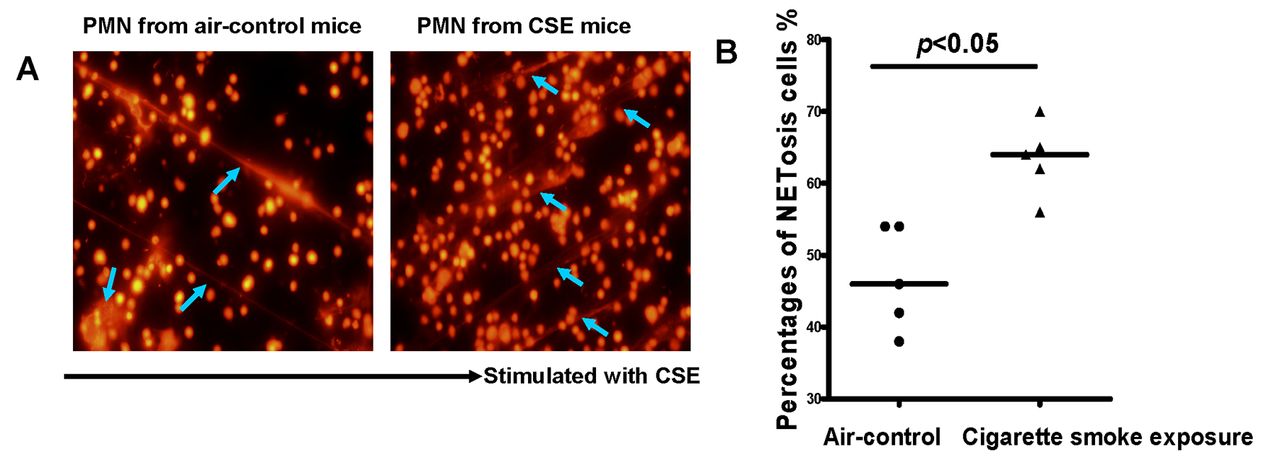
Cigarette smoke extract (CSE) directly triggered the formation of neutrophil extracellular traps (NETs) in vitro. (A) Neutrophils isolated from peripheral blood of air-control (left panel) or cigarette smoke exposed mice (right panel) were stimulated for NET formation with CSE (0.3%) at 37°C for 4 hours. Neutrophils were then fixed with 4% PFA, stained with propidium iodide (PI) and then examined by fluorescence microscopy (NETs: blue arrows); original magnification, ×40. (B) Comparisons of percentages of NETosis cells in response to CSE between two groups. The data are representative of five separate experiments. Data are expressed as medians. The comparisons were determined by Mann-Whitney test.
NETs induced by CSE-activated pDCs
Previous studies have suggested that the immunogenic complexes composed of antimicrobial peptides (LL37) and self-DNA, both major components of NETs, can efficiently trigger pDC activation in patients with SLE. To test whether CSE-induced NETs could activate pDCs, primary pDCs isolated from air-control mice were stimulated with AluI-derived NETs or MNase-derived NETs which were produced by the stimulation of CSE in vitro. Eighteen hours after pDC stimulation, we found that both AluI-derived NETs and MNase-derived NETs induced by CSE efficiently drove pDC maturation, as shown by the upregulated surface expression of CD40, CD86 and MHC-II(figure 5A–F). This was accompanied by a rapid secretion of IFN-α, IL-6 and IL-12/p70 (figure 5G–I). Moreover, this proinflammatory effect of NETs was dependent in part on the amount of nuclease-derived NETs (data not shown). Interestingly, we observed that pDCs exposed to CSE and NETs simultaneously, a situation more usual in vivo, led to an increased expression of CD40, CD86 and MHC-IIas well as a significant high production of IFN-α, IL-6 and IL-12/p70 compared with unstimulated pDCs (figure 5D–I). We also achieved similar results with air-control mice in cigarette smoke exposed mice (see online Supplementary figure S2).

Neutrophil extracellular traps (NETs) induced by cigarette smoke extract (CSE) activated pDCs. Primary plasmacytoid dendritic cells (pDCs) were isolated from spleens of air-control mice by negative selection with Plasmacytoid Dendritic Cell Isolation Kit (Miltenyi Biotec). Primary pDCs were then incubated with serum-free medium alone or treated with MNase-derived NETs (50 ng/mL) or Alu I-derived NETs (50 ng/mL) or Alu I-derived NETs (50 ng/mL) combined with CSE in serum-free medium for 18 hours at 37°C in 5% CO2. The maturation of pDCs was determined by evaluating MHC class II and costimulatory molecule CD40, CD86 expression. The concentrations of interleukin (IL)-6, IL-12/p70 and interferon (IFN)-α in supernatant were determined by ELISA. Representative flow cytometric histograms of (A) CD40, (B) CD86 and (C) MHC-II on pDCs in each group. Comparisons of proportions of (D) CD40+, (E) CD86+ and (F) MHC-II+pDCs in each group. Comparisons of concentrations of (G) IL-6, (H) IL-12/p70 and (I) IFN-α in each group. All data are representative of five independent experiments. Data are expressed as medians. The comparisons were determined by Kruskal-Wallis one-way ANOVA on ranks. *p<0.05, NS, not significant.
pDCs activated by CSE-induced NETs primed naïve CD4+ T cells to differentiate Th1 and Th17 cells
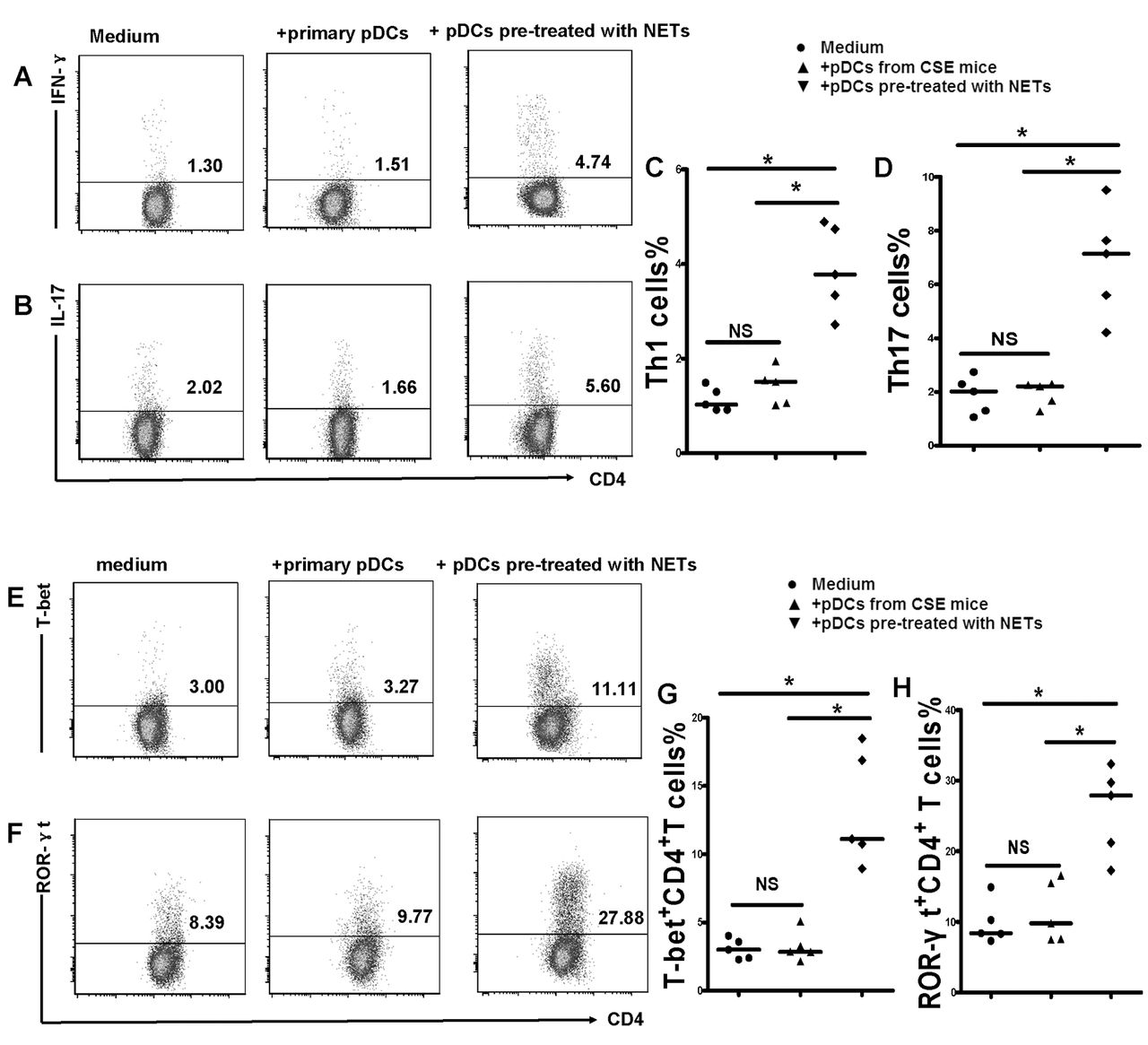
The chronic activation of pDCs may subsequently elicit an antigen-specific T-cell response. Thus, we isolated naïve CD4+ T cells from the spleens of air-control mice and cocultured them with pDCs from cigarette smoke exposed mice or NET-activated pDCs for 4 days in vitro. The results demonstrated that naïve CD4+ T cells cocultured with pDCs from cigarette smoke exposed mice or NET-activated pDCs can be differentiated into Th1 and Th17 patterns that have a higher level of IFN-γ and IL-17 compared with those cocultured with primary pDCs from air-control mice (figure 6) (see online supplementary figure S3). We also found that T-bet and ROR-γt, the specific transcription factor of Th1 and Th17, was significantly increased in CD4+ T cells cocultured with pDCs from cigarette smoke exposed mice or NET-activated pDCs (figure 6) (see online supplementary figure S3). These observations collectively indicate that pDCs activated by CSE-induced NETs could induce a Th cell mediated immune response in vitro, an event that is important to the pathogenesis of COPD (figure 7).
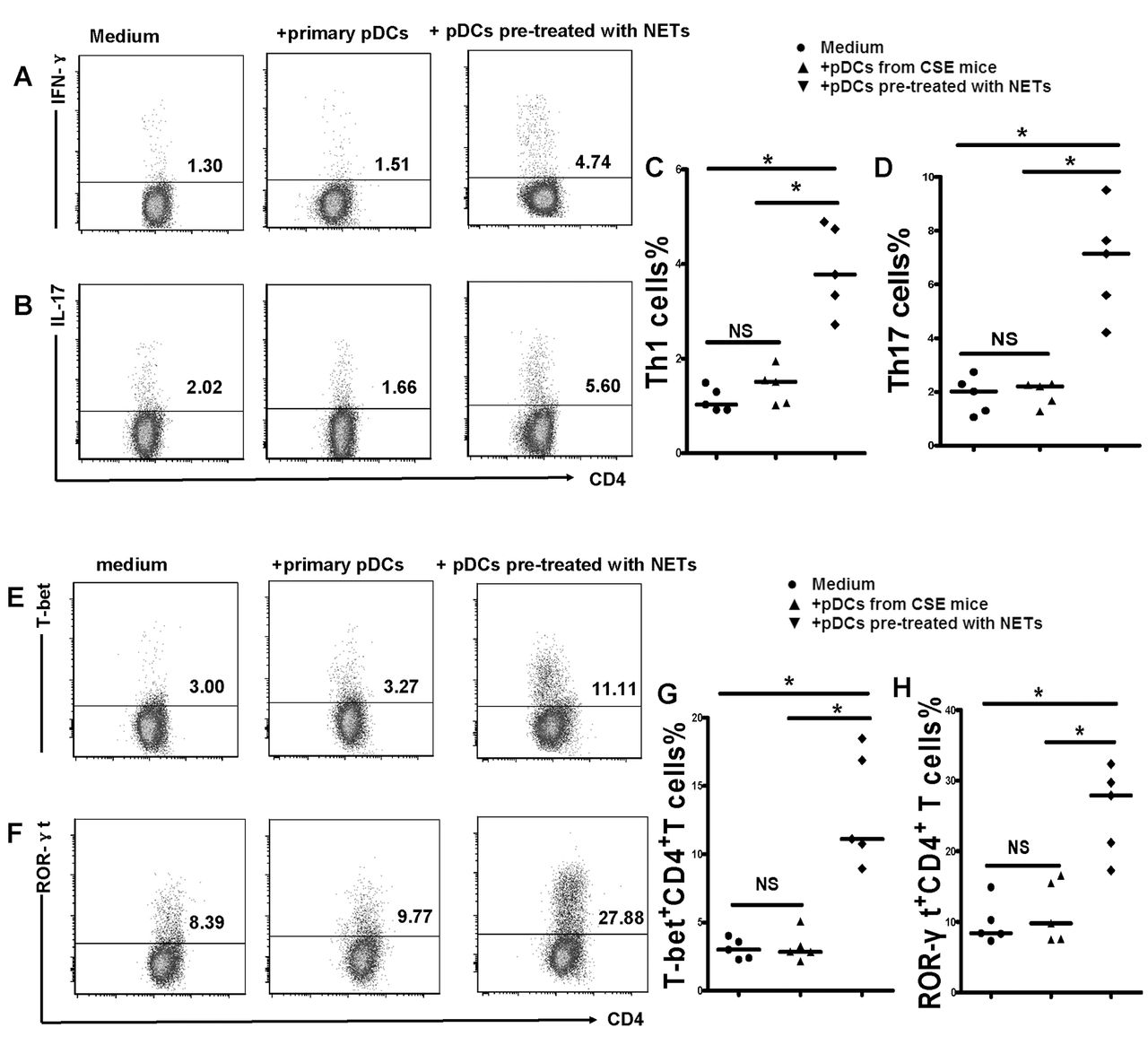
Plasmacytoid dendritic cells (pDCs) activated by cigarette smoke extract (CSE)-induced neutrophil extracellular traps (NETs) were sufficient to promote naïve CD4+ T cells differentiated into T helper 1 (Th1) and Th17 cells. Naïve CD4+ T cells isolated from spleens were cocultured with primary pDCs or pDCs pretreated with CSE-induced NETs (1:1 ratio) in 24-well plates. In some experiments, naïve CD4+ T cells were cultured in 24-well plates precoated with antimouse CD3 mAbs (5 ug/mL; Ebioscience) and soluble anti-CD28 (2 μg/mL; Ebioscience) and were supplemented with interleukin (IL)-2 (10 ng/mL; Ebioscience) on day 2. Four days later, the cells were harvested and stained for surface marker PerCP-CD4 followed by intracellular labelling of PE-IL-17 and APC-IFN- after 25 ng/mL PMA and 1 ug/mL ionomycin activation for 4 hours in the presence of GolgiStop. Alternatively, cells were stained for surface marker PerCP-CD4 followed by intracellular labelling of PE-T-bet and APC-ROR-γt after being permeabilised with Foxp3/Transcription Factor Staining Buffer Set. The expression of IL-17 and interferon (IFN)-γ was determined by flow cytometry. Representative flow cytometric dot plots of (A) Th1 and (B) Th17 cells in each group. Comparisons of proportions of (C) Th1 and (D) Th17 in each group. Representative flow cytometric dot plots of (E) T-bet and (F)ROR-γt in each group. Comparisons of proportions of (G) T-bet and (H) ROR-γt in each group. All data are representative of five independent experiments. Data are expressed as medians. The comparisons were determined by Kruskal-Wallis one-way ANOVA on ranks. *p<0.05, NS, not significant.
{kind=link}
{kind=link}
{kind=link}
![[SP1.jpg]](https://thorax.bmj.com/content/thoraxjnl/72/12/1084/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
![[SP2.jpg]](https://thorax.bmj.com/content/thoraxjnl/72/12/1084/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
![[SP3.jpg]](https://thorax.bmj.com/content/thoraxjnl/72/12/1084/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
{kind=link}
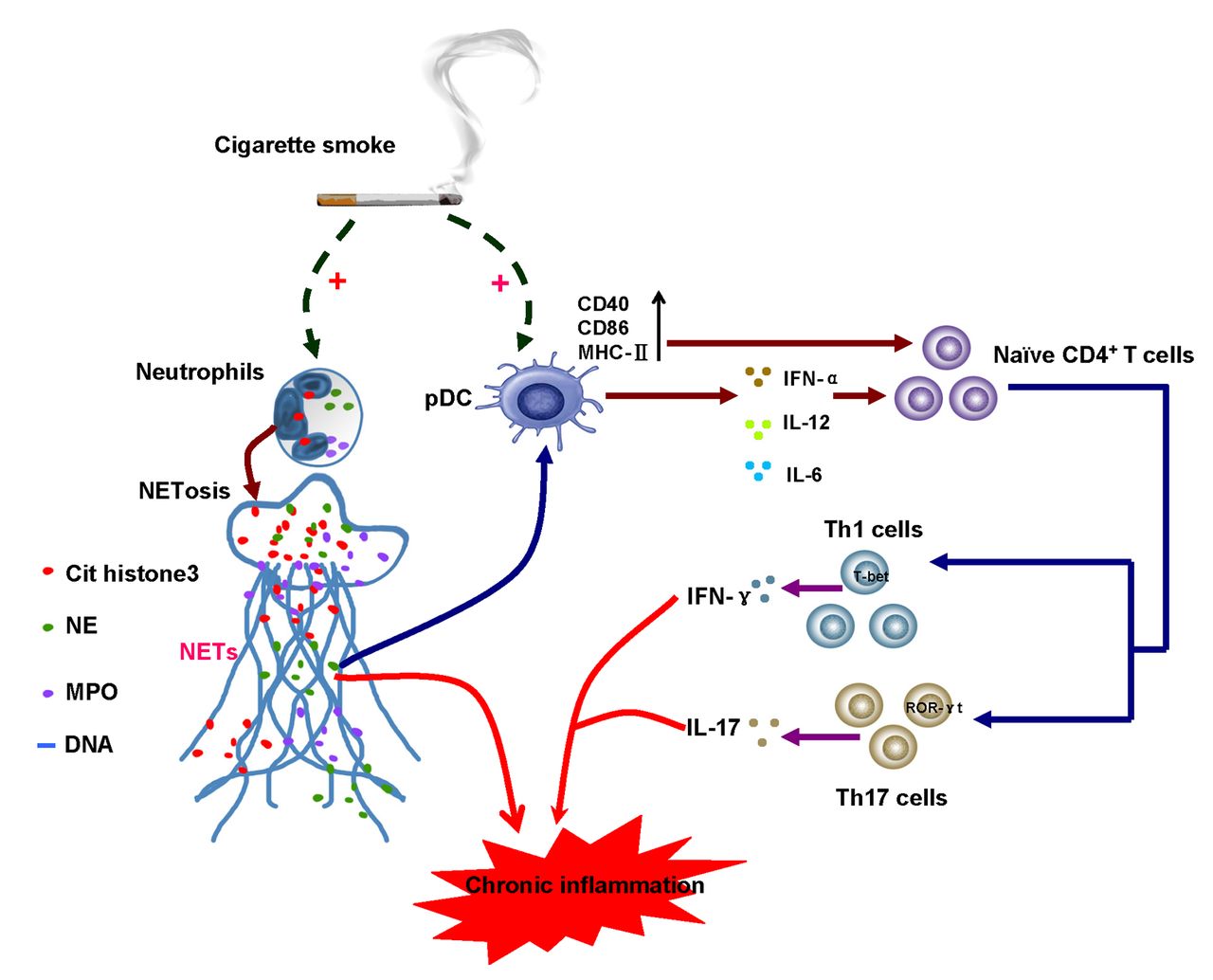
A Proposed model of crosstalk between neutrophil extracellular trap (NET) formation, plasmacytoid dendritic cells (pDCs) activation and the initiation of T helper (Th)-cell-mediated response in the context of chronic cigarette smoke exposure. Cigarette smoke triggers neutrophils to undergo NETosis. The dsDNA, proteins or enzymes within NETs could directly induce tissue injury, or alternatively, promote pDC maturation and activation. pDCs treated with cigarette smoke extract (CSE)-induced NETs were shown to develop a proinflammatory phenotype characterised by the upregulation of MHC-IIand costimulatory molecules CD40 and CD86, and the release of proinflammatory cytokines interferon (IFN)-α, interleukin (IL)-12 and IL-6. Through cell–cell contact or cytokine production, activated pDCs were sufficient to prime naïve CD4+ T cells to differentiate into Th1 or Th17 linage, thus orchestrating the Th-cell-mediated immune response and contributing to the chronic inflammation in the context of chronic cigarette smoke exposure.
Discussion
A central question in the current inflammatory diagram of COPD is how the normal response of the host to cigarette smoke alters into an amplified, pathological inflammatory injury; more importantly, how does chronic cigarette smoke exposure provoke the adaptive immune response from the innate immune system. As a major component of the innate immune system and the main source of elastase, neutrophils have long been recognised to play a crucial role in smoking-related emphysema. Nevertheless, this map of neutrophils remains incompletely described in detail. Here our results demonstrate that neutrophils were more susceptible to form NETs but were defective in clearance within the context of chronic cigarette smoke exposure, which was tightly related to the sustained activation of pDCs. CSE may represent a previously unrecognised facilitator that triggers neutrophils to undergo NETosis in vitro. Furthermore, CSE-induced NETs served as an endogenous danger signal potentially driving pDC maturation and activation, thereby initiating T-cell-mediated local and/or systemic inflammation as seen in smoking-related COPD.
The initial reports described NETs as a barrier efficient in trapping, confining and eliminating invading microorganisms, which serves as an important complement to the antimicrobial armamentarium of neutrophils. Despite the beneficial role of NETs in bacterial infections, emerging evidence has revealed that NETs and NET-associated proteins exert a deleterious effect on the host and are implicated in vasculitis, sterile inflammation, atherosclerosis and thrombosis, transfusion-related acute lung injury, primary graft dysfunction after lung transplantation and tumour metastasis.20–25 Limited clinical observations have found a massive presence of NETs and NETotic neutrophils in the sputum of patients with cystic fibrosis and COPD, which strongly suggests a role of NETs in chronic airway diseases.5 6 26 In most cases, the formation of NETs is a highly regulated process because of the very short lifespan of neutrophils and the rapid clearance of NETs. It is well known that cigarette smoke elicits a state of oxidative stress in which large amounts of ROS are produced. Interestingly, NETosis is a process largely dependent on the production of ROS.27 In the present study, we observed that PMNs from cigarette smoke exposed mice produced more NETs in response to PMA compared with air-control mice. Our finding is in line with a recent study which found that neutrophils from patients with rheumatoid arthritis displayed enhanced NET formation.28 The quick removal of NETs may represent a remedial strategy that has evolved to prevent excessive NETs, which are harmful to the host. Several studies have shown that serum from a subgroup of patients with SLE who had more active disease displayed impaired function in degrading NETs, partly due to the presence of DNase I inhibitors or anti-NET antibodies.29 30 Consistent with these observations, we observed that serum from our smoking mouse model of emphysema was also defective in the clearance of NETs. Although the precise mechanisms underlying the inability to degrade NETs in the serum from cigarette smoke exposed mice are still unclear, the imbalance between NET generation and clearance may aggravate inflammatory damage and increase the risk of an autoimmune response to NET components.
The observation that NETs were also abundant in the sputum of healthy smokers who did not exhibit airflow limitation further indicates that NETs may play a harmful role in the absence of microbial stimuli.5 Accordingly, the current study found that CSE appeared to be a potent inducer that directly promoted the formation of NETs in vitro, and CSE-induced NETs shared identical characteristics to PMA-induced NETs in some conserved proteins (data not shown). Even though NETs are present in the sputum of most smokers, it is unclear if or how cigarette smoke induced NETs cause irreversible lung damage in some smokers that is a feature of COPD.
In contrast to the traditional notion of neutrophils becoming rapidly exhausted in a course of acute inflammation, emerging evidence now proposes that these cells survive much longer than original suggested and may play a substantial role in shaping the adaptive immune response in chronic inflammation.11 Until recently, neutrophils were found to be capable of modulating the maturation and activation of DCs, and in turn, the proliferation and polarisation of antigen-specific T cells.11 However, the specific role of NETs in the initiation of the adaptive immune response in cigarette smoke related emphysema has not yet been characterised. In agreement with published data,16 we observed that pDCs appeared to be fully activated in mice after chronic cigarette smoke exposure, which was associated with pathogenic Th1 and Th17 responses. pDCs form a highly sensitive sentinel network to sense external or internal danger signals by rapidly mounting a robust type 1 IFN response to infections, or on the other hand, initiating adaptive immunity. Thus, the disturbance of pDC functions may lead to unregulated immune responses. In the present study, we found that CSE-derived NETs functioned as a principal activator effectively promoting pDC maturation and subsequently eliciting an aberrant release of IFN-α, IL-6 and IL-12 in vitro. These results are in line with the findings from patients with SLE in which anti-ribonucleoprotein (RNP) IgG–treated neutrophils are able to induce pDCs to secrete IFN-α, IL-6 and tumour necrosis factor α.31 We also found that NET-activated pDCs were able to promote the generation of Th1 and Th17 responses in coculture with naïve CD4+ T cells. Contrary to common views that mature pDCs hold tolerogenic properties in peripheral tissue,32 our observations suggest that pDCs activated by CSE-derived NETs are more likely to exhibit an aggressive phenotype sufficient to initiate pathologic Th1 and Th17 responses in vitro. COPD is a highly heterogeneous disease, thus the translation of the acute inflammatory information contained in NETs into a chronic, self-perpetuating inflammatory process mediated by a specific T-cell response by pDCs may partly explain why only some smokers develop COPD and why the inflammation persists even after smoking cessation.
Some limitations in this study need to be taken into consideration. First, all the mice used for the experiments were male; thus, the generalisability of the results is limited. Second, we used PMA to evaluate the general susceptibility of PMNs from cigarette smoke exposed mice to NET formation in parts of this study. Nevertheless, the relevance of these non-physiological stimuli to the pathology of the lung need to be considered as more physiological or pathological stimuli were not used and other investigations have reported pathogenic bacteria and its associated components (lipopolysaccharide in particular), inflammatory mediators and endogenous damage-associated molecular patterns can all trigger NET formation. Lastly, although the circulating PMNs and spleen pDCs used in this study shared some similar characteristics with those of the lung and may highlight the systemic feature of smoking-related emphysema, how well these blood and spleen cells equate to a lung disease requires further determination.
In conclusion,we have demonstrated that NET-mediated pDC activation may represent a critical link between innate and adaptive immune responses under conditions of chronic inflammation induced by cigarette smoking. These findings suggest that targeting NETs may offer a novel therapeutic approach to smoking-related COPD.
References
Footnotes
Contributors Study design: XNZ and SLQ. Data acquisition and statistical analyses: SLQ, HZ, QYT, JB, ZYH, JQZ, MHL, MCD, JMD and GNL. Manuscript writing: SLQ and XNZ.
Funding This work was supported by grants from the National Natural Science Foundation of China (No. 81660007, No. 81260013 and No. 81260011).
Competing interests None declared.
Ethics approval Medical Ethical Committee of the First Affiliated Hospital of Guangxi Medical University.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves