Article Text

Abstract
Rationale The role bacteria play in the progression of COPD has increasingly been highlighted in recent years. However, the microbial community complexity in the lower airways of patients with COPD is poorly characterised.
Objectives To compare the lower airway microbiota in patients with COPD, smokers and non-smokers.
Methods Bronchial wash samples from adults with COPD (n=18), smokers with no airways disease (n=8) and healthy individuals (n=11) were analysed by extended-culture and culture-independent Illumina MiSeq sequencing. We determined aerobic and anaerobic microbiota load and evaluated differences in bacteria associated with the three cohorts. Culture-independent analysis was used to determine differences in microbiota between comparison groups including taxonomic richness, diversity, relative abundance, ‘core’ microbiota and co-occurrence.
Measurement and main results Extended-culture showed no difference in total load of aerobic and anaerobic bacteria between the three cohorts. Culture-independent analysis revealed that the prevalence of members of Pseudomonas spp. was greater in the lower airways of patients with COPD; however, the majority of the sequence reads for this taxa were attributed to three patients. Furthermore, members of Bacteroidetes, such as Prevotella spp., were observed to be greater in the ‘healthy’ comparison groups. Community diversity (α and β) was significantly less in COPD compared with healthy groups. Co-occurrence of bacterial taxa and the observation of a putative ‘core’ community within the lower airways were also observed.
Conclusions Microbial community composition in the lower airways of patients with COPD is significantly different to that found in smokers and non-smokers, indicating that a component of the disease is associated with changes in microbiological status.
- Bacterial Infection
- COPD Pathology
- Opportunist lung infections
- Respiratory Infection
Statistics from Altmetric.com
Key messages
What is the key question?
Is the lower airway microbiota of patients with COPD different from that observed in ‘healthy’ smokers and healthy non-smokers?
What is the bottom line?
For the majority of patients with COPD, the lung microbiota is significantly different from that present in people without any marked pulmonary disease.
Why read on?
Using culture-dependent and culture-independent methods, we describe the microbiota of the lower airways in disease and health. The study further extends understanding of the underlying relationship between bacterial colonisation and progression of lung disease in conditions such as COPD.
Introduction
COPD is characterised by irreversible airflow obstruction, mucus hypersecretion, small airway fibrosis and destruction of alveolar spaces resulting in emphysema.1
Risk factors for bacterial colonisation/infection of the airways include smoking, severity of airway obstruction and/or inflammation of the airways.2 Moreover, bacterial colonisation/infection of the lower airways may play an important role in the progression of COPD lung disease,3–8 with studies demonstrating an association between the presence of bacteria and greater levels of airway inflammation, exacerbation frequency and decline in lung function.2 ,9–11 The precise function bacteria play in the development of COPD lung disease has been difficult to define in part due to previous assumptions regarding the sterile nature of the lungs in healthy individuals.12–16 Recent findings from culture-dependent and culture-independent studies show that the lungs are not sterile in healthy subjects.7 ,17 Confirmation of the non-sterile nature of the respiratory tract in healthy individuals provides us with a better understanding regarding the complex nature of bacterial community structures within lower airways. Furthermore, it enables an assessment of how bacterial community structures in healthy individuals differ from those in patients with COPD, and how these differences might drive inflammation and disease progression.
This study is the first to simultaneously use bronchial wash samples obtained from patients with COPD, smokers and non-smokers without any pulmonary morbidities to determine airway microbial composition using both culture-dependent and culture-independent methodologies. We hypothesised that the airways of patients with COPD harbour a distinct microbiota, providing a disease-associated microbial signature.
Materials and methods
Ethics statement and patient recruitment
The study was approved by the Office for Research Ethics Northern Ireland (ORECNI, REC reference number: 09/NIR03/42) with all participants providing written informed consent prior to procedures being carried out. The COPD subjects were recruited from patients listed for bronchoscopy for clinical indications at the Belfast City Hospital. The ‘healthy’ subjects volunteered to the study and were recruited from the general population. All participants were involved in a study to determine the effects of cigarette smoke on airway epithelial cells, which has been previously published.18 Participants were divided into three groups: (1) clinically stable patients with COPD (n=18) with mild to severe airflow obstruction according to the published ‘Global Strategy for Diagnosis, Management, and Prevention of COPD’ guidelines (GOLD 2009; http://www.goldcopd.org/), (2) smoking volunteers (‘Healthy’ Smokers, HS; n=8) and 3) non-smoking volunteers (Healthy Volunteers, HV; n=11). All COPD study subjects underwent a CT scan with radiographic findings consistent with a diagnosis of COPD alone for all subjects. In particular, there was no evidence of bronchiectasis, interstitial lung disease or lung carcinoma.
Sample collection and processing
During elective transoral fiberoptic bronchoscopy, bronchial washing was performed through a bronchoscope (TeleMed Systems, Massachusetts, USA) in the third-generation bronchus of the right middle lobe, with specimens obtained by flushing the area under investigation with 30 mL of sterile saline. This anatomical site was selected in order to maximise return. Approximately 20 mL was retrieved on each occasion with relatively minor individual variability. Washings were taken prior to any other procedures in the study. Findings on the CT scan did not influence the site of sample collection.
Culture-based investigation of bronchial wash samples and detection of isolated bacteria were performed as previously described19 with minor modifications as highlighted in the online supplementary material. For culture-independent analysis, an aliquot of bronchial wash was retained for analysis by Illumina MiSeq sequencing as described below.
Supplemental material
Molecular detection—Illumina MiSeq sequencing
16S rRNA marker-gene sequencing was performed on the Illumina MiSeq platform at the Wellcome Trust Sanger Institute (Hinxton, UK). We used the V1–V2 region as it covers a hyper-variable section of the 16S rRNA gene, and is relatively short, meaning that it can be effectively covered using Illumina MiSeq paired end sequencing. Moreover, it has previously been shown to be useful in 16S rRNA marker-gene surveys.20–22 After normalisation, on the basis of the sample with the fewest reads (9347), a total of 345 839 reads were included in all downstream analysis. Following initial quality filtering, sequences were clustered into their representative operational taxonomic units (OTUs) based on 97% sequence similarity. Previously, Salter and colleagues highlighted the importance of monitoring potential spurious sequences originating from DNA-contaminated consumables used in molecular studies.23 In the current study we stringently monitored potential background amplicon reads found in the negative control. This was then followed by the removal of the relevant OTUs that traversed both the negative control and clinical samples. All OTUs that were considered to have been caused by technical contamination during DNA extraction or library preparation were removed prior to further downstream analysis. Further information regarding library preparation, sequencing, as well as data handling and processing is provided in the online supplementary material.
Intrasample similarities/differences were assessed through sample richness (number of counted taxa), community evenness and diversity (Shannon–Wiener index). Relatedness between samples was assessed through principal coordinates analysis of weighted UniFrac (phylogenetic) values between COPD and combined ‘healthy’ cohorts, with the axes scaled by the percentage of the variance that they contained. The amount of variation explained was evaluated for the first three principal components. Furthermore, potential overlap of taxa within communities was assessed through the construction of co-occurrence/coexclusion networks as detailed in the online supplementary material.
The relationship between the main ecological indexes of the metacommunity (ie, taxonomic richness, Shannon–Wiener index of diversity, evenness, dominance) and lung function (FEV1% of predicted) was assessed by performing regression analysis (coefficients of determination (adjusted R2); residuals; significance) as well as by calculating the correlation (Spearman's rs; significance) between variables.
Statistical analysis
A detailed description of the statistical methods is provided in the online data supplementary material. The sample size was determined by the primary study.18
For culture-dependent analysis, total viable counts (TVCs) of aerobic and anaerobic bacteria within each group were compared using the Wilcoxon signed rank test, with comparison of aerobic, anaerobic and total bacterial load between the three cohorts performed by the Kruskal–Wallis test. Similarly, for culture-independent analysis, comparison of community richness and diversity between the three groups was assessed by the Kruskal–Wallis test, with post hoc tests performed using the Mann–Whitney test with Bonferroni adjustment applied. Categorical variables, such as gender and inhaled steroid usage, were compared using the Pearson's χ2 test with Yates’ continuity correction.
For comparison of the relative contribution of different phyla and genera, we applied Metastats (http://metastats.cbcb.umd.edu/detection.html),24 with 999 permutations, which allowed identification of differentially abundant features between particular taxa within the three cohorts. Resulting p values that indicated statistical significance were adjusted according to the Bonferroni approach allowing for multiple testing correction. Further information on the Metastats method is included in the online supplementary material.
Overall agreement beyond chance alone between methodologies in detecting the absence or presence of particular taxa was estimated using Cohen's κ coefficient as described in the online supplementary material.
Significance testing between COPD versus the combined health-associated cohorts for principal coordinates analysis was assessed using the ANOSIM (Analysis of Similarity) function as implemented in the QIIME (Quantitative Insights Into Microbial Ecology) pipeline.25 Co-occurrence between bacterial taxa was assessed using Spearman's rank correlation coefficient with a further definition of the ‘core’ microbiota as described in the online supplementary material.
All statistical analyses and graphical representations were carried out in Graphpad Prism (V.5.00) and the R environment (http://www.r-project.org). Further details regarding specific packages and analysis are provided in the online supplementary material.
Results
Patient demographics
Patient demographic data are summarised in table 1. The majority of subjects in the COPD group were referred for bronchoscopy after an episode of haemoptysis for exclusion of an underlying malignancy. None of the participants in this group were on antimicrobial therapy for at least 8 weeks prior to sample collection and no individual had bronchodilator reversibility of greater than 10%. Seventeen (94%) patients with COPD received long-acting β-agonist (LABA) and inhaled corticosteroid (ICS) treatment. Patients with COPD were classified as having mild obstruction with FEV1 >80% predicted (n=1); moderate obstruction with FEV1 50%–79% predicted (n=11) and severe obstruction with FEV1 30%–49% predicted (n=6). Percent FEV1 of predicted was significantly different between the three groups (p<0.0001; Kruskal–Wallis test) (table 1). Further analysis between each of the cohorts demonstrated a significant difference in FEV1% predicted between COPD and both HS and HV (p=0.0003 and p<0.0001, respectively; Kruskal–Wallis test with Bonferroni correction). There was no significant difference between HS and HV (p=0.231; Kruskal–Wallis test with Bonferroni correction).
Study demographics and sputum culture of PPM
Culture-dependent analysis
Bacteria were cultured in high abundance (up to 2.14×107 cfu/mL) in bronchial wash samples from all patients with COPD. A mean (range) of 7 (4–15) different taxa were cultured per sample with a median (range) TVC of 6.22×104 (1.36×103–2.14×107) cfu/mL bronchial wash, with aerobic bacteria detected in significantly higher numbers than anaerobic bacteria (p<0.001, Wilcoxon signed rank test; figure 1). Bacteria were also detected in high abundance (up to 1.01×107 cfu/mL) in 7/8 HS. The seven culture-positive HS samples had a mean (range) of 9 (5–11) different taxa cultured per sample with a median (range) TVC of 1.39×105 (2.24×104–1.01×107) cfu/mL bronchial wash. There was no significant difference between the TVC of aerobic and anaerobic bacteria (p=0.127, Wilcoxon signed rank test; figure 1). Similarly, bacteria were cultured in high abundance (up to 4.21×106 cfu/mL) in bronchial wash samples from 9/11 HV. In the nine culture-positive HV samples, there was a mean (range) of 11 (5–17) different taxa with a median (range) TVC of 4.04×105 (3.38×103–4.21×106) cfu/mL bronchial wash. Aerobic bacteria were significantly more abundant than anaerobic bacteria (p<0.001; Wilcoxon signed rank test). There was no significant difference in the total load of both aerobic and anaerobic bacteria between the three cohorts (p=0.264 and p=0.313, respectively, Kruskal–Wallis test; figure 1). Further information regarding culture of potential pathogenic microorganisms (PPMs) and other bacteria is shown in table 1 and online supplementary table S1, respectively.

Total viable counts (cfu/mL) of aerobic and anaerobic bacteria cultured from bronchial wash samples collected from stable patients with COPD (n=18), ‘healthy’ smokers (HS) (n=8) and healthy volunteers (HV) (n=11). *Comparing two groups using Wilcoxon signed rank test; ^comparing three groups using Kruskal–Wallis test.
Culture-independent analysis
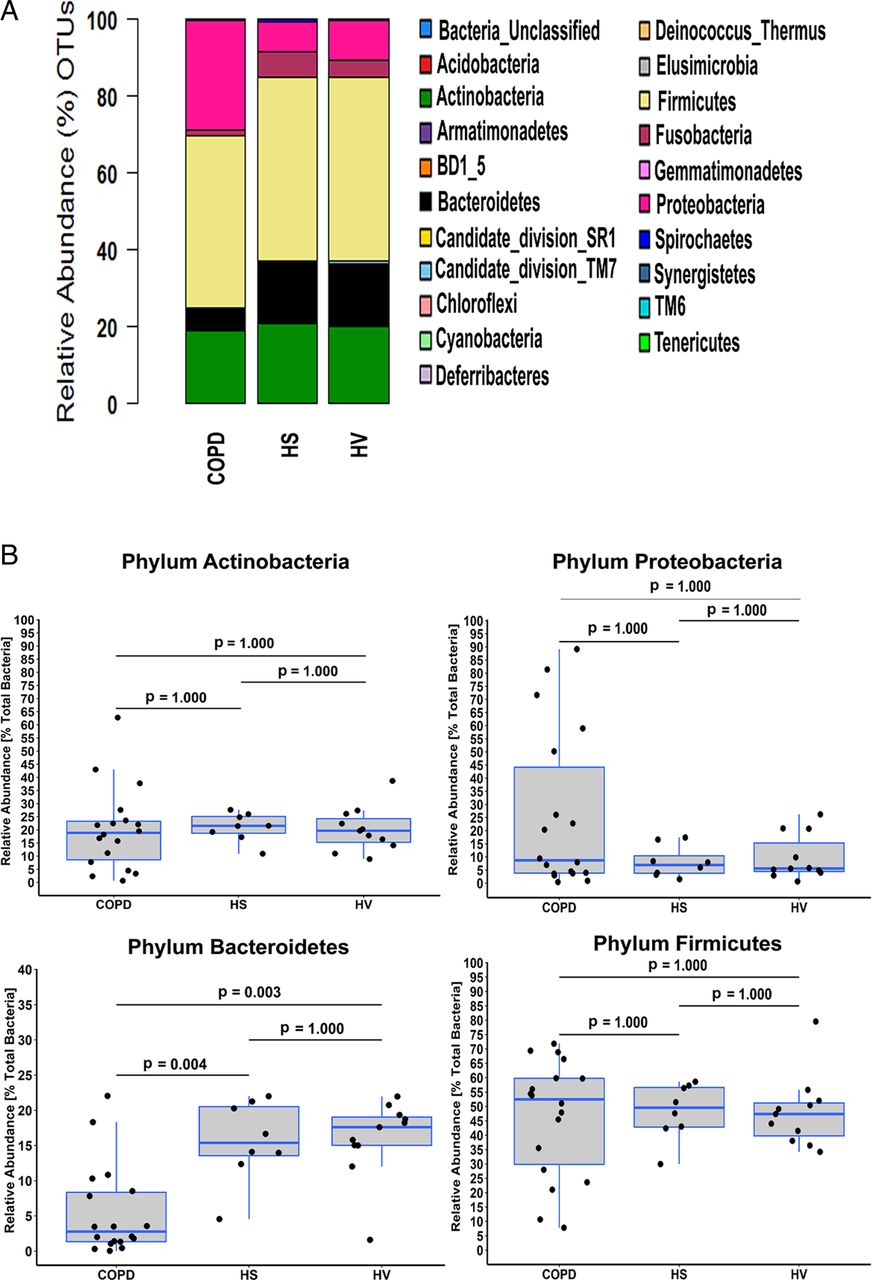
In total, we observed 332 different taxa in the three cohorts. However, a minority of taxa (n=19) accounted for more than 90% of the total amplicon reads (figure 2A). To assess how four of the main phyla contributed to the observed differences, we compared their relative abundances in the three sample cohorts (figure 2B). No differences were apparent for Actinobacteria, Firmicutes and Proteobacteria. However, significant differences were apparent for Bacteroidetes (p=6.56×10−4, Kruskal–Wallis test) with post hoc tests revealing that abundance was significantly greater in both HS and HV compared with patients with COPD (p=0.003 and p=0.004, respectively, Fisher's exact test, Bonferroni adjustment; figure 2B); no difference was observed between HS and HV (p=1.000, Fisher's exact test, Bonferroni adjustment; figure 2B).

Taxonomic identification at phylum level in each cohort. (A) Stacked bar representing the relative abundance of phyla accounting for the total sequence count in the three cohorts. (B) Comparison of the relative abundance of the Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria phyla between the three cohorts. Values shown depict percentage relative abundance of the total bacteria detected within each individual. Horizontal lines represent the median value. Statistical analysis for comparing groups was performed using Fisher's exact test in Metastats. Multitest corrections were performed using Bonferroni adjustment. OTUs, operational taxonomic units.
At the genus level, differences were apparent in community composition between COPD, HS and HV (figure 3A). The three main anaerobic genera, Prevotella, Actinomyces and Veillonella, contributed proportionally less to the total relative abundance in COPD (13%) compared with either HS (30%) or HV (29%). In contrast, Haemophilus spp. accounted for 12% of the total sequence reads in COPD, compared with 5% and 4% in HS and HV, respectively. Streptococcus spp. contributed equally to all three cohorts, accounting for 35% (COPD), 30% (HS) and 30% (HV) of the total relative abundance. When we compared relative abundances in the three sample cohorts, significant differences were only apparent for Prevotella and Pseudomonas (p=0.001 and p=0.006, respectively; Kruskal–Wallis test). Post hoc tests revealed that for Pseudomonas, proportional abundance was significantly greater in COPD compared with HS and HV patients (p=0.009 and p=0.040, respectively; Fisher's exact test, Bonferroni adjustment; figure 3B). However, the overall relative contribution was low in all three cohorts, with only three COPD samples demonstrating the relative abundance of Pseudomonas spp. above 1%, with a single COPD sample out of those three accounting for the majority of sequence reads assigned to this genus (>80% of the total sequences in that sample). Furthermore, this sample was the only one that grew Pseudomonas aeruginosa by extended culture, and identification of the relevant OTUs through NCBI BLASTN (https://blast.ncbi.nlm.nih.gov/) indicated that it belonged to P. aeruginosa. However, after removing this sample as an outlier, we still observed a significant difference in the mean Pseudomonas spp. abundance between COPD and both HS and HV (0.42%, 0.03% and 0.04%, respectively; p=0.006; Kruskal–Wallis test). In contrast, for Prevotella, abundance was significantly less in COPD compared with HS and HV patients (p=0.006 and p=0.007, respectively; Fisher's exact test, Bonferroni adjustment; figure 3B). Further and more detailed analyses of the observed differences between the three cohorts are provided in the online supplementary material.
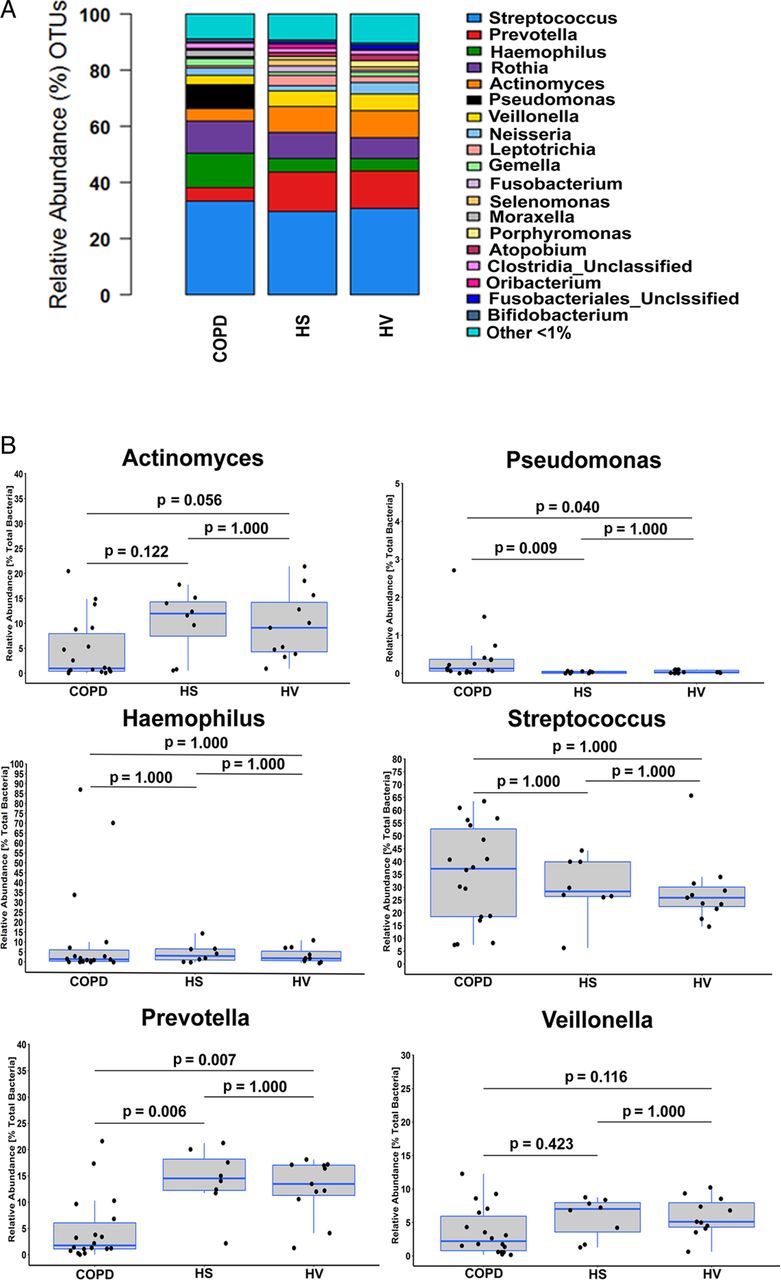
Taxonomic identification at genus level in each cohort. (A) Stacked bar representing the relative abundance of genera accounting for the total sequence count in the three cohorts. (B) Comparison of the relative abundance of a number of the main genera: Actinomyces, Haemophilus, Prevotella, Pseudomonas, Streptococcus and Veillonella between the three cohorts. Values shown depict percentage relative abundance of the total bacteria detected within each individual. The group ‘Other <1%’ denotes taxa that did not reach at least 1% of total read count in any individual sample, respectively. Horizontal lines represent the median value. Statistical analysis for comparing groups was performed using Fisher's exact test in Metastats. Multitest corrections were performed using Bonferroni adjustment on significant p values. OTUs, operational taxonomic units.
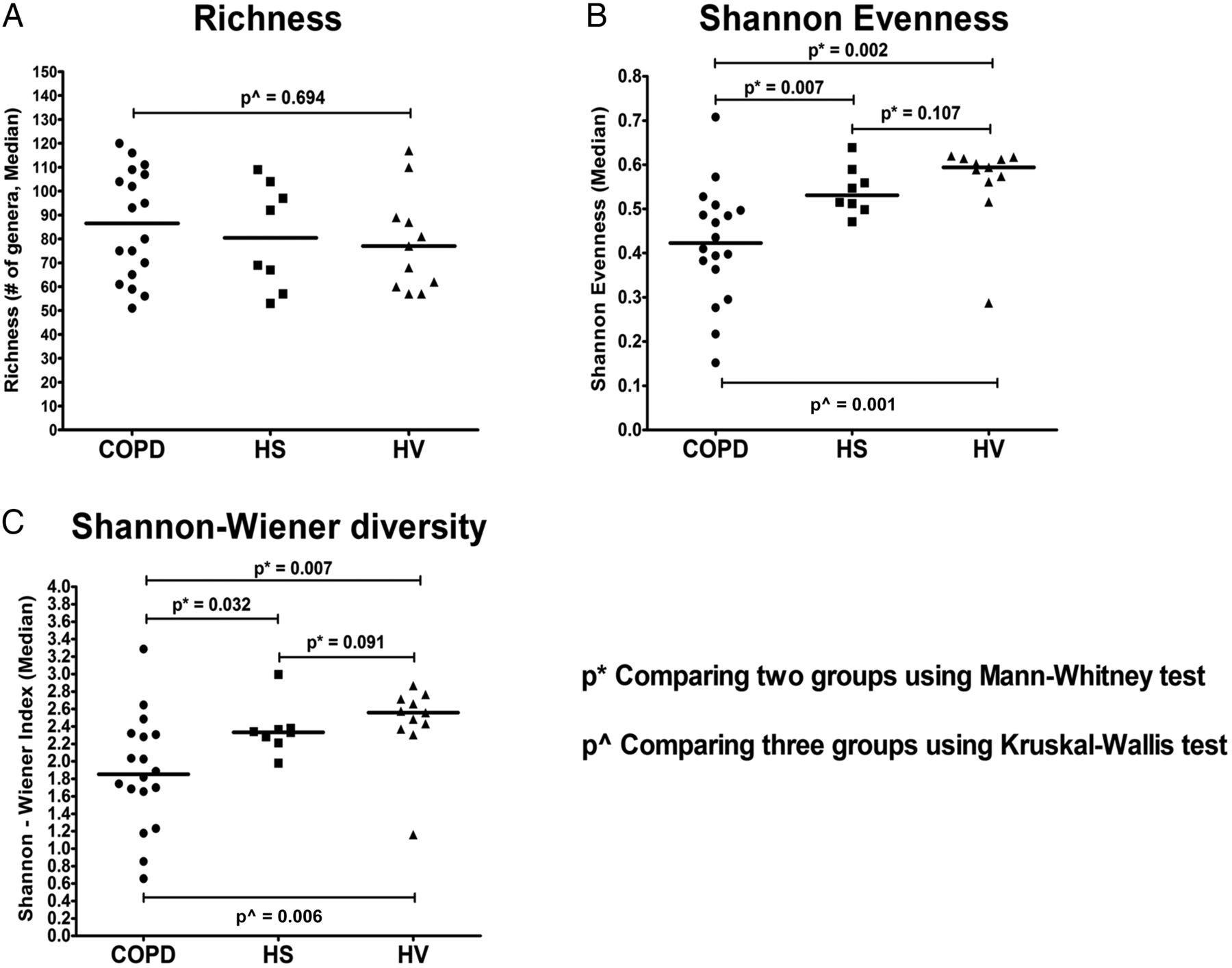
Comparison of taxonomic richness (ie, the number of taxa present in a sample at a particular taxonomic level) demonstrated no significant difference in observed taxa between COPD, HS and HV (figure 4A; p=0.694, Kruskal–Wallis test). However, community evenness (ie, the degree that taxa present in a particular sample are of equal proportions or relative abundance) was significantly different between the three sample cohorts (figure 4B, p=0.001; Kruskal–Wallis test), with lower evenness observed in COPD compared with both HS and HV (p=0.007 and p=0.002, respectively; Mann–Whitney test, Bonferroni adjustment). Similarly, diversity (Shannon–Wiener index), which takes into account both richness and evenness within the community, was also significantly different between communities (figure 4C, p=0.006; Kruskal–Wallis test). Post hoc tests revealed that diversity was lower in COPD compared with both HS and HV (p=0.032 and p=0.007, respectively; Mann–Whitney test, Bonferroni adjustment). No differences were apparent for either evenness or diversity between HS and HV (p=0.091, Mann–Whitney test, Bonferroni adjustment) (figure 4C).

Taxonomic richness (A), taxonomic evenness—Shannon evenness (B) and Shannon–Wiener index of diversity (C). The horizontal black line represents the median (50th quartile). *Comparing two groups using Mann–Whitney test with Bonferroni adjustment; ^comparing three groups using Kruskal–Wallis test. p<0.05 denotes statistical significance. HS, ‘healthy’ smokers; HV, healthy volunteers.
We also compared agreement between culture-dependent and culture-independent detection of the main bacterial taxa according to Cohen's κ coefficient. There was significant agreement for the main taxa detected by either culture-dependent or culture-independent method (see online supplementary table S3).
Principal coordinate analysis with weighted UniFrac distance was performed to assess relatedness between samples from COPD and the ‘healthy’ comparison groups (HS and HV). These groups were combined due to the strong similarities between these communities with regard to taxa distribution, community evenness, taxonomic diversity and measurement of lung function. The first two principal components accounted for 72.1% of variance explained and there was a broad clustering effect separating the corresponding communities based on which cohort they originated from (ANOSIM, R=0.311, p=0.001, 999 permutations). The majority of the HS and HV subjects were in the ‘healthy’ cluster with few subjects from the COPD cohort aligning within this cluster. The COPD subjects that aligned towards the ‘healthy’ cohort had higher taxonomic richness and diversity, generally associated with lower age and better lung function, compared with the rest of the COPD cohort (see online supplementary figures S1 and S4).
We also investigated the putative structural background of the bacterial community in patients with COPD compared with the ‘healthy’ comparison group. First, we assessed the overall correlating taxa in all 37 samples (see online supplementary figure S2). In general, potential clinically relevant taxa, such as Haemophilus spp., Streptococcus spp. and Pseudomonas spp., did not correlate with other members of the community. We further compared putative co-occurrence/coexclusion between taxa in COPD and combined HS and HV (see online supplementary figure S2). A number of obligate anaerobic bacteria, such as members of the genera Prevotella, Veillonella, Actinomyces and Atopobium, formed the main connected component in both networks highlighting the role anaerobic bacteria may play as ‘core’ community members of the lower airways. Taxa conventionally considered as PPMs (ie, Pseudomonas spp. and Haemophilus spp.) in COPD did not correlate with any other taxa, indicating a stronger dominance and difference in habitat adaptation within their corresponding community. Furthermore, assigning specific OTUs detected in >90% of samples and accounting for more than 1% total sequence read abundance to the ‘core’ microbiota demonstrated that it consisted of those same anaerobic taxa, in addition to members of the Streptococcus and Rothia genera (see online supplementary figure S3 and table S4).
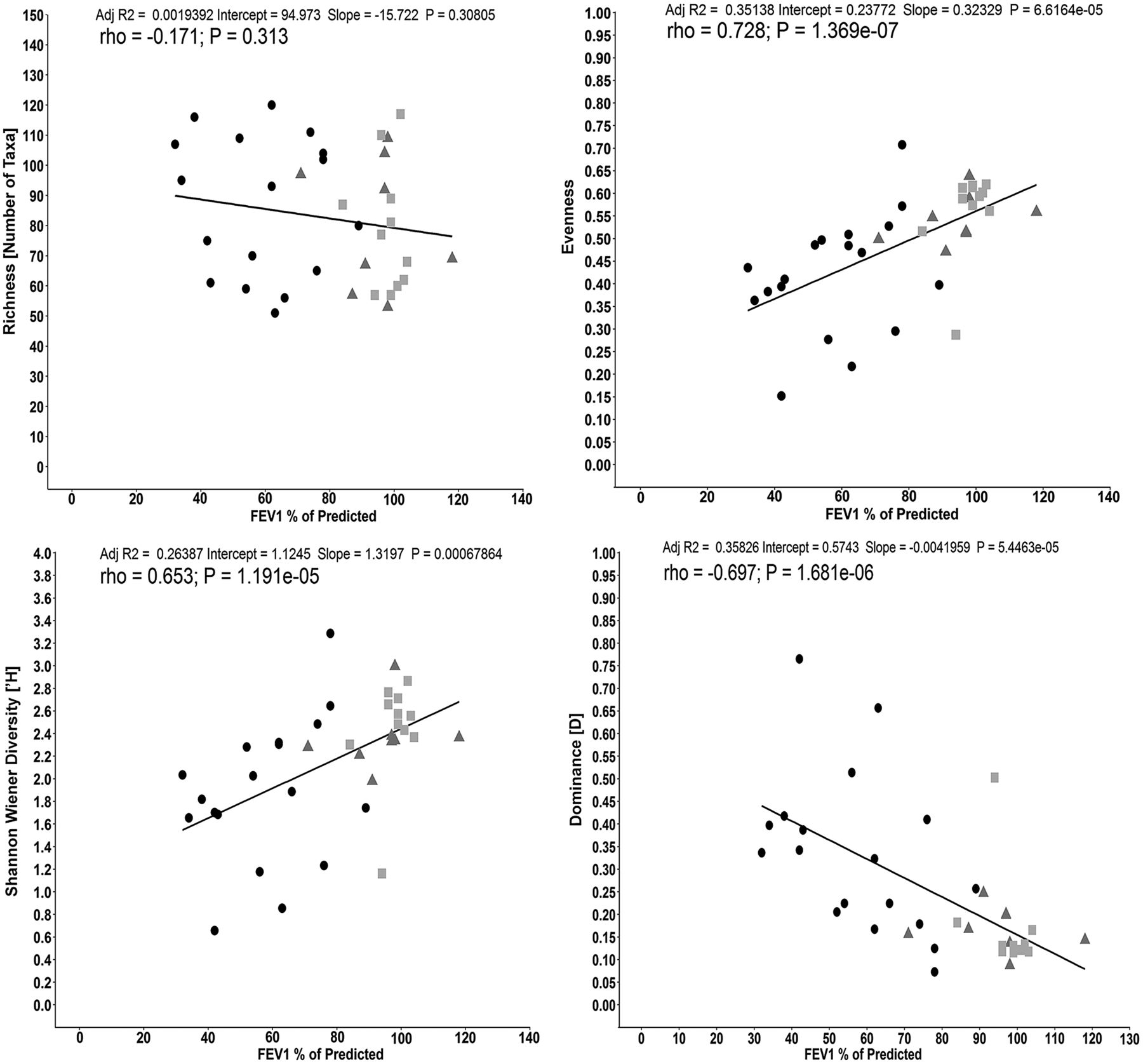
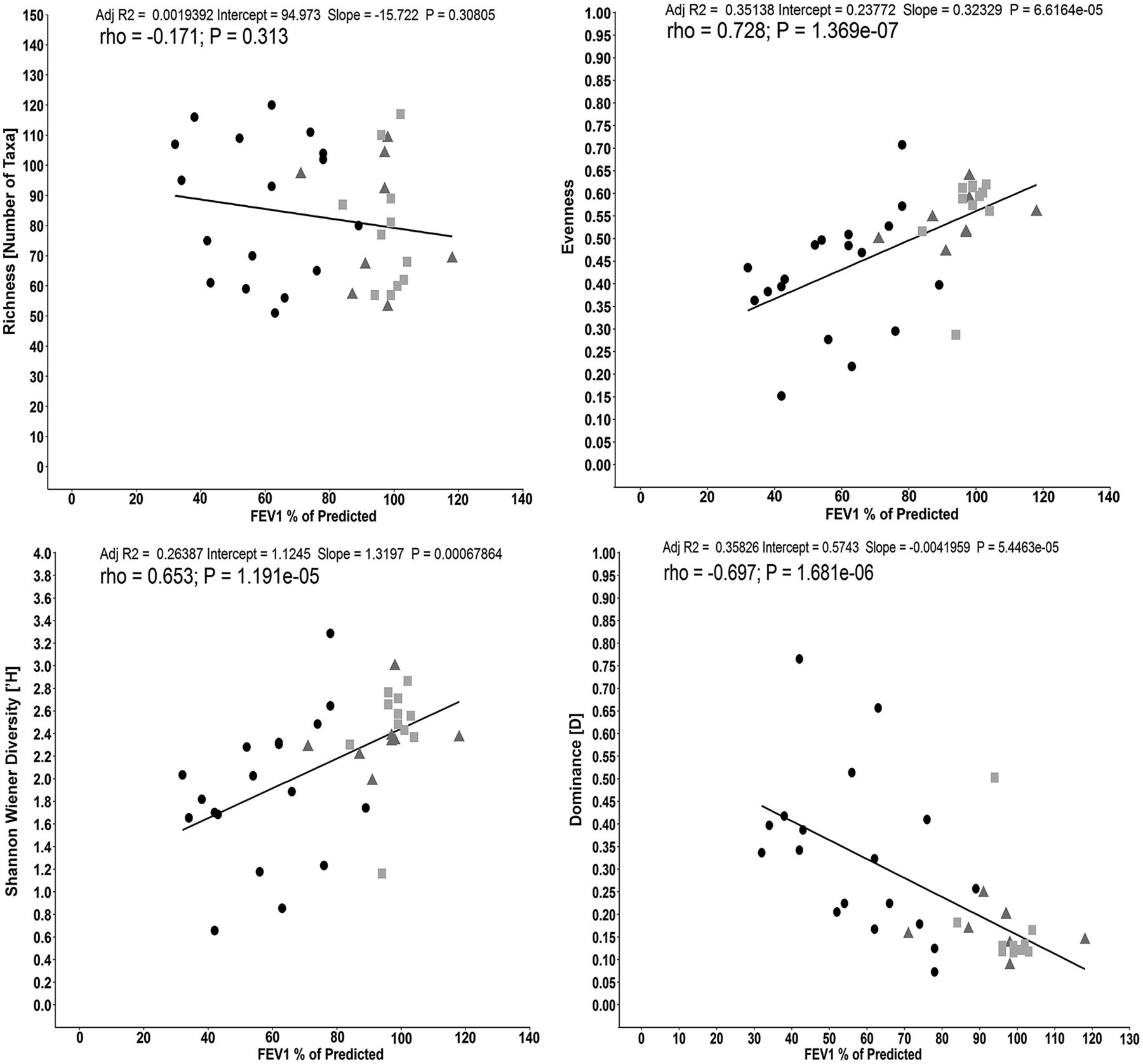
To investigate the relationship between taxa richness, diversity, evenness, dominance and lung function presented as FEV1 of % predicted within the meta-community (all three cohorts), we performed regression and correlation analysis (figure 5A–D, respectively). Taxonomic richness was not associated with changes in lung function (adjusted R2=0.002, p=0.308; r=−0.171, p=0.313). However, both diversity (Shannon–Wiener index) and evenness demonstrated a positive relationship (adjusted R2=0.264 (p=6.786×10−4) and adjusted R2=0.351 (p=6.616×10−5), respectively) and correlation (r=0.653 (p=1.191×10−5) and r=0.728 (p=1.369×10−7), respectively) with higher lung function. There was also a strong negative relationship between taxa dominance and lung function (adjusted R2=−0.358 (p=5.446×10−5) and r=−0.697 (p=1.681×10−6)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between lung function (FEV1% of predicted) for (A) taxonomic richness, (B) Shannon–Wiener diversity, (C) evenness and (D) dominance within the whole metacommunity (n=37; COPD:  ; Smokers (‘healthy’ smokers, HS):
; Smokers (‘healthy’ smokers, HS):  ; Non-smokers (healthy volunteers, HV):
; Non-smokers (healthy volunteers, HV):  ). Line indicates linear regression and the Spearman's correlation coefficient (rs) is shown (p<0.05 denotes statistical significance).
). Line indicates linear regression and the Spearman's correlation coefficient (rs) is shown (p<0.05 denotes statistical significance).
Discussion
This is the first study employing bronchial washes with both extended-culture and culture-independent (Illumina MiSeq amplicon sequencing) methods to compare the bronchial microbiota of patients with COPD and two comparison groups (HS and HV). By culture-dependent analysis, we detected both aerobic and anaerobic bacteria in significant numbers in samples from all three cohorts, with significant agreement between methodologies in the detection of the main bacterial genera observed.
We did not observe significant differences in community richness between COPD and either of the two comparison groups, but the overall community diversity and evenness was significantly lower for patients with COPD. Community dominance of a single taxa, reported in other chronic respiratory diseases such as cystic fibrosis (CF)26 and bronchiectasis,27 ,28 was rarely observed in the COPD cohort, with the exception of three samples in which clinically relevant taxa (P. aeruginosa, n=1 patient; Haemophilus influenzae, n=2 patients) were detected by culture. In general, single taxa dominance did not exceed 50% of the observed relative abundance within a sample. This apparent lack of single taxa dominance may be explained by the fact that our cohort was not established based on individuals known to be colonised by certain microorganisms that have been frequently associated with chronic infection in respiratory diseases including COPD.
Our findings contrast with a number of published studies that reported an increase in bacteria belonging to the Firmicutes phylum during most stages of COPD lung disease,3 ,8 ,29 ,30 while Erb-Downward and colleagues observed the predominance of the Proteobacteria phylum in patients with advanced disease.7 However, all our patients with COPD were stable at the time of sampling and there was a good spread in airflow obstruction that may account for some of the differences observed with previously published studies. Our findings demonstrate no difference between COPD and the ‘healthy’ comparison groups with respect to the proportional contribution of Firmicutes or Actinobacteria between the three cohorts. For Proteobacteria, we observed a trend towards differences between COPD and either of the comparison cohorts, and it is possible that in a larger study such differences might become more prevalent. However, Bacteroidetes, which includes a number of common anaerobic bacteria, for example Prevotella, contributed significantly less to the relative abundance in COPD when compared with either of the ‘healthy’ cohorts. Furthermore, analysis of the community structures within the three cohorts at a higher taxonomic resolution (ie, at the genus level) demonstrated an increased relative contribution by Pseudomonas spp. in patients with COPD compared with either of the ‘healthy’ cohorts. However, this was mainly attributed to three COPD samples, which accounted for the majority of sequence reads belonging to Pseudomonas spp., with one of those showing to be positive for P. aeruginosa both by extended-culture and by molecular detection. The other main Pseudomonas spp. detected at a higher relative abundance in COPD subjects were identified as being non-P. aeruginosa. The relative proportion of Haemophilus spp. was observed to be similar between all three groups. Conversely, our observation that a number of common anaerobic bacteria, such as Prevotella, Veillonella and Actinomyces, were reduced in COPD compared with the comparison groups may point to a beneficial or ‘normal state’ role for these bacteria. Ordination, as displayed through principal coordinates analysis, demonstrated a broad separation of communities into COPD and ‘healthy’ groups. However, a number of subjects from the COPD cohort were aligned within the ‘healthy’ cluster. Taxonomic richness and diversity were generally higher in these subjects who had lower age and better lung function compared with the rest of the COPD cohort, highlighting the diverse nature of chronic lung disease such as COPD. Co-occurrence of bacterial taxa and the observation of a putative ‘core’ community within the lower airways, including Prevotella spp., Veillonella spp. and Actinomyces spp., were also apparent in this study. This is similar to observations we have previously made in other chronic airways disease such as CF and non-CF bronchiectasis, and in a number of sites within the upper airways of healthy individuals.31 However, correlation between taxa, as described here, does not indicate a functional relationship between members of the community. Therefore, further studies, focusing on the functional capabilities such taxa display within the complex community assembles found in the lower airways, are required to determine if anaerobic bacteria contribute to symbiosis in health and/or disease.
The observed relationship between lower airway microbial community characteristics and lung function, as a marker of disease severity, is in agreement with previous findings for other lung diseases characterised by chronic infection such as CF and bronchiectasis.32–36 Furthermore, this highlights how narrowing of bacterial community composition, in the context of microbial diversity and taxa dominance, may be linked to clinical markers of disease progression.
Our methodological approach has a number of limitations. First, the current study included a relatively small number of subjects. However, to date, this is one of the largest studies investigating the bronchial microbiota simultaneously in subjects with COPD, smokers and non-smokers using samples obtained by bronchoscopy. Second, we did not attempt to differentiate between viable and non-viable cells prior to culture-independent analysis. Previous studies have suggested that this may result in overestimation of the underlying diversity though the inclusion of genomic DNA from non-viable microbial cells.37 However, none of the COPD subjects were under active antimicrobial therapy; therefore, the relative bias of non-viable cells within the COPD cohort would be minimal when compared with either of the ‘healthy’ cohorts. Third, using bronchial wash may also affect the sensitivity of culture-dependent recovery of bacteria due to the significant dilutional effect of injecting up to 30 mL of sterile saline. Collecting samples from the lower airways by bronchoscopic procedures can also potentially introduce the risk of contamination from the upper airway microbiota. However, the majority of techniques used to sample the lower airways traverse the oro-pharynx and the mouth with an associated risk of oropharyngeal contamination. Our protocols were standardised for sample collection allowing comparison across the three sample cohorts.
A number of studies have suggested that the use of ICSs and inhaled bronchodilators may alter the microbiota. For example, Pragman et al29 demonstrated changes in the lung microbiota related to use of both ICSs and inhaled bronchodilators with clustering of ICS and LABA users. Furthermore, Huang et al3 observed a trend towards greater diversity in samples from patients using ICS with enrichment for Proteobacteria and members of other phyla. In the present study, 17 of the 18 patients with COPD were prescribed both ICS and LABA therapy, whereas the HS and HV cohorts were naive to both treatments. Therefore, the potential impact of both ICS and LABA treatment on the lung microbiota could not be assessed due to the lack of an appropriate control group, such as corticosteroid-‘naive’ COPD subjects.
In conclusion, our results show dynamic bacterial communities within the airways of individuals with COPD, as well as those not affected by chronic airways disease. Furthermore, microbial community composition in COPD is significantly different to that in both smokers and non-smokers, indicating that a component of the disease is associated with changes in microbiological status. There is a clear need for larger cohort studies to assess how bacterial community composition affects patients with COPD. Moreover, data from such studies may inform better targeted treatment for patients, improved long-time prognosis and quality of life.
Acknowledgments
The authors wish to thank David Harris, Paul Scott, Alan Walker and the Wellcome Trust Sanger Institute's Illumina Miseq sequencing team for generating the 16S rRNA gene sequence data.
References
Footnotes
GGE, MMT and JSE are joint senior authors on this paper.
Twitter Follow Gisli Einarsson at @geinarsson
Contributors ME, JSE., GGE and MMT conceived and designed research; DMC collected samples and clinical data; GGE and DMC performed research; GGE and MMT analysed data; GGE performed bioinformatic analysis; JP, ME and JSE made intellectual contributions; GGE, MMT and JSE wrote the paper. All authors read and approved the final version of the manuscript.
Funding GGE, MMT, LM and JSE were supported through a US-Ireland Project Partnership Grant by the HSC Research and Development, Public Health Agency, Northern Ireland. GGE, MMT and JSE were supported through CFMATTERS, a Seventh Framework Programme Project No. 603038. Funding for JP and 16S rRNA gene sequencing was provided by the Wellcome Trust (grant 098051). MMT was supported by a Health and Social Care Research and Development, Public Health Agency, Northern Ireland, funded UK National Institute for Health Research Career Scientist Award. DMC and ME were supported through the Northern Ireland Chest Heart and Stroke (NICHS Project 2010_10) and the Northern Ireland Research and Development Office (REF: EAT/3978/08).
Competing interests None declared.
Ethics approval Office for Research Ethics Northern Ireland.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data deposition: Illumina MiSeq sequence data have been submitted to the NCBI Sequence Read Archive (SRA) under the study accession number SRP057611 http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?study=SRP057611.