Article Text

Abstract
Cystic fibrosis (CF) is a life-limiting disease characterised by recurrent respiratory infections, inflammation and lung damage. The volume and composition of the airway surface liquid (ASL) are important in maintaining ciliary function, mucociliary clearance and antimicrobial properties of the airway. In CF, these homeostatic mechanisms are impaired, leading to a dehydrated and acidic ASL. ASL volume depletion in CF is secondary to defective anion transport by the abnormal cystic fibrosis transmembrane conductance regulator protein (CFTR). Abnormal CFTR mediated bicarbonate transport creates an unfavourable, acidic environment, which impairs antimicrobial function and alters mucus properties and clearance. These disease mechanisms create a disordered airway milieu, consisting of thick mucopurulent secretions and chronic bacterial infection. In addition to CFTR, there are additional ion channels and transporters in the apical airway epithelium that play a role in maintaining ASL homeostasis. These include the epithelial sodium channel (ENaC), the solute carrier 26A (SLC26A) family of anion exchangers, and calcium-activated chloride channels. In this review we discuss how the ASL is abnormal in CF and how targeting these alternative channels and transporters could provide an attractive therapeutic strategy to correct the underlying ASL abnormalities evident in CF.
- Cystic Fibrosis
Statistics from Altmetric.com
Airway surface liquid
The airway surface liquid (ASL) is a thin layer of fluid covering the luminal (apical) surface of the airway epithelium and plays a key role in airway homeostasis. The volume, pH, ionic and nutrient content of the ASL are all important in regulating antimicrobial activity, ciliary function and mucociliary transport (figure 1). Antimicrobial factors found within the ASL are involved in innate and adaptive host defence mechanisms that protect the airway from inhaled pathogens.
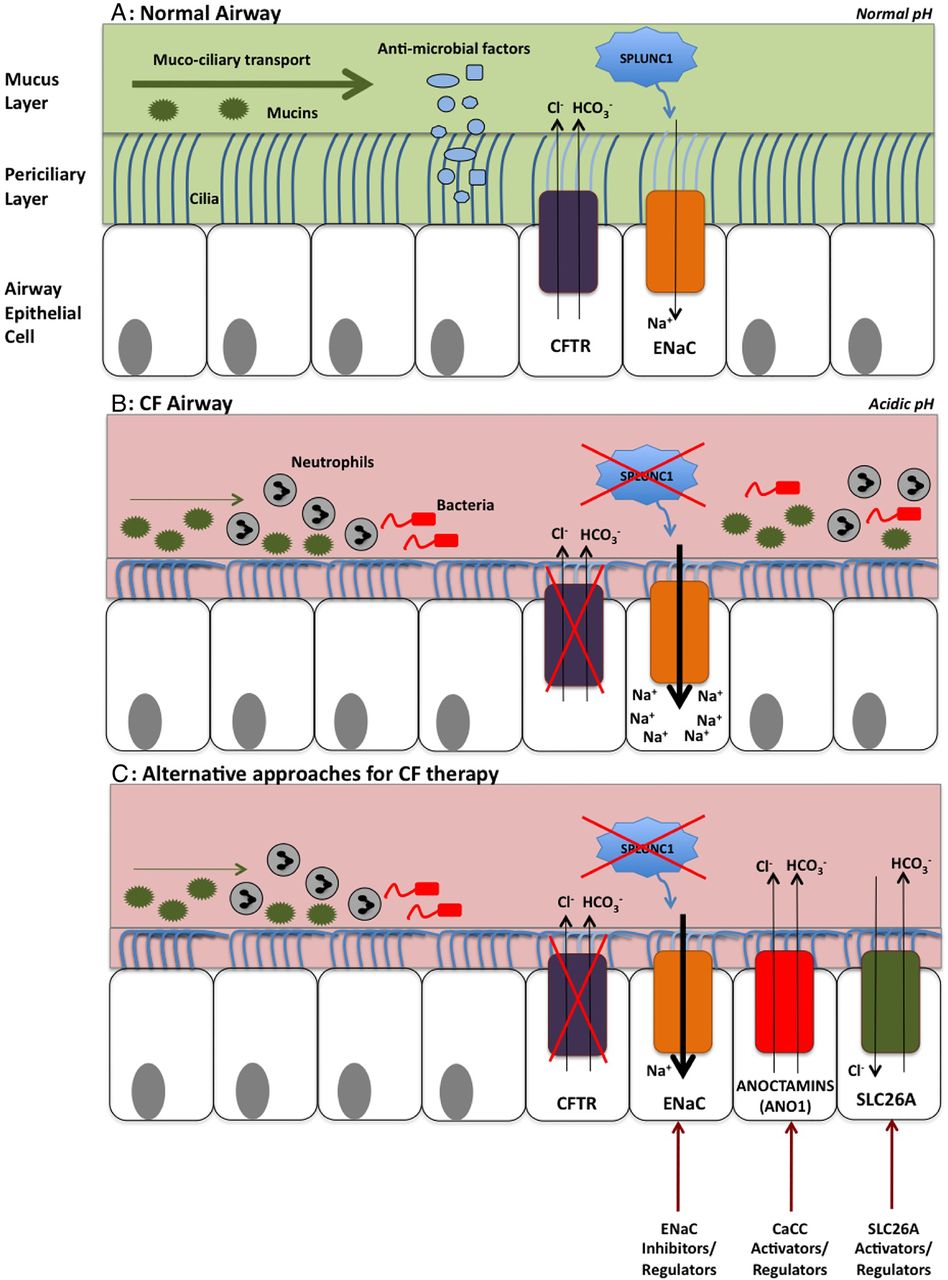
{kind=link}
The airway surface liquid (ASL) in cystic fibrosis (CF) and potential targets for therapies. (A) The normal airway: the ASL consists of a mucus layer containing large gel-forming mucins that trap inhaled particles for removal by mucociliary clearance. High concentrations of osmotically active tethered mucins within the periciliary layer (PCL) form a tight macromolecular mesh and preserve PCL volume. The CF transmembrane conductance regulator protein (CFTR) transports chloride and bicarbonate into the airway. Sodium is absorbed via the epithelial sodium channel (ENaC), contributing to ASL hydration and mucociliary transport. SPLUNC1 is found in abundance within the ASL and regulates ASL volume by inhibiting ENaC. The ASL also contains antimicrobial factors, which play a key role in innate host defence. (B) The CF airway: defective chloride and bicarbonate transport by abnormal CFTR leads to a dehydrated and acidic ASL. SPLUNC1 fails to regulate ENaC, leading to sodium hyper-absorption and further ASL volume depletion. The acidic environment reduces antimicrobial function and increases mucus viscosity. These mechanisms result in the production of thick, mucopurulent secretions and bacterial colonisation. In this dehydrated state, water preferentially moves out of the mucus layer, increasing its concentration. The PCL volume is initially preserved, but eventually reaches a critical threshold at which water also exits the PCL. The mucus layer subsequently compresses the PCL, compromising ciliary activity and mucociliary clearance. (C) Alternative approaches for CF therapy: targeting other apical channels and transporters may be potential alternative avenues for CF therapy. ENaC inhibition or regulation could help to preserve ASL volume. Regulation or activation of alternative apical chloride channels, including the anion exchanger solute carrier 26 A (SLC26A) family, and the calcium-activated chloride channel, anoctamin 1, could correct defective apical chloride transport and therefore the ASL abnormalities evident in CF lung disease.
Alterations in ASL composition and disruption of this tightly regulated environment have been linked to the pathogenesis of cystic fibrosis (CF) lung disease. Knowledge of this area has progressed rapidly of late and the focus of this review is to highlight how factors involved in maintaining ASL homeostasis could be potential targets for novel therapeutic strategies in people with CF.
The ASL consists of two distinct layers (figure 1). The mucus layer contains large gel-forming mucins which trap inhaled particles for removal from the lung by mucociliary clearance. Beneath this is the periciliary layer (PCL), which lies adjacent to the airway epithelial cells and surrounds the cilia. High concentrations of large membrane-bound mucins within the PCL are tethered to the cilia and surface of apical epithelial cells.1 They form a tight macromolecular mesh, preventing penetration of mucus and inhaled particles into the PCL. The greater osmotic modulus generated by these membrane mucins allows relative preservation of PCL volume in a healthy hydrated airway.1
This recent ‘gel-on brush’ ASL model has enabled greater understanding of how disruption of this tightly regulated system can alter mucus clearance and contribute towards pathogenesis in airways diseases.1
The ASL is dehydrated in CF
In CF, dysfunctional CF transmembrane conductance regulator (CFTR) protein leads to defective apical chloride and bicarbonate transport into the airway lumen. Subsequent sodium and water compartmental shifts, in addition to this defective anion transport, result in a dehydrated airway surface and the production of viscous, acidic, muco-purulent secretions that are difficult to clear.
In a dehydrated airway environment, water preferentially moves out of the ASL mucus layer, increasing its concentration and osmotic modulus. Eventually, in severely dehydrated CF airways, a critical threshold is reached whereby water also exits the PCL. The mucus layer subsequently compresses the PCL, affecting ciliary activity and therefore mucociliary transport.1 In CF, this leads to mucus stasis, airway infection, inflammation, and progressive lung disease (figure 1).
The ASL pH is reduced in CF
Defective CFTR-mediated bicarbonate transport leads to an acidic ASL, which creates an unfavourable environment for the function of many antimicrobial factors. A reduction in ASL pH in the CF pig model has been shown to decrease antimicrobial activity, which was restored by directly increasing pH.2 Bicarbonate is also required for maintaining normal mucus properties and homeostasis. Its depletion results in dense, impenetrable mucus that remains tethered to the epithelium, thus impairing mucociliary transport.3
Therapeutic targets for ASL regulation in CF
The underlying genetic defect in CF is well defined, and recent therapeutic strategies have been directed towards correcting the function of mutant CFTR protein.4 For example, the CFTR potentiator, ivacaftor, has proved clinically effective in the 5% of people with the Gly551Asp-CFTR gating mutation.5 In 2015 the results of phase III trials of the combination of ivacaftor and the corrector lumacaftor in patients homozygous for the most frequent CF-causing mutation Phe508del were published, but showed less clinically significant benefits.6 The combination drug received US Food and Drug Administration approval in 2015 for treatment in patients who are Phe508del homozygotes. However, this approach will not benefit all patients; around 50% are not Phe508del homozygotes, and alternative strategies to correct the underlying fluid and pH imbalance in the ASL are required.
Stimulation of other channels and transporters involved in apical ion transport may compensate for the abnormalities in ion transport, solute trafficking, pH and ASL volume that are seen with the absence of functional CFTR. These ‘by-pass therapies’ could provide complementary or even alternative therapies in CF that would benefit all patients.
ENaC
The epithelial sodium channel, ENaC, plays a key role in apical sodium absorption and is important in regulating ASL hydration and mucociliary transport. ENaC activity is upregulated in CF, which leads to sodium hyper-absorption and contributes to ASL dehydration (figure 1).7 ,8
The precise underlying mechanisms for ENaC upregulation in CF are not fully understood but knowledge in this area is increasing. CFTR dysfunction is thought to be linked to an increase in ENaC activity, however this has not been demonstrated consistently in all experimental models and their relationship remains in question.9
Furthermore, ENaC activation occurs through a number of other mechanisms, including the cleavage of its subunits by endogenous serine proteases.10 One such example, neutrophil elastase, is abundant in inflamed CF airways and in other diseases including COPD, which may be contributory to ENaC activation.11 ,12
Given the hyperactivity of ENaC in CF, inhibitory agents have been investigated as potential therapies. In particular, amiloride has previously failed to show sustained clinical improvements due to low potency and rapid airway absorption.13 Other potential agents, including derivatives of amiloride, have since been developed and undergone phase I and II clinical trials. However, challenges have been faced with developing new compounds that provide adequate lung delivery without unwanted pulmonary and systemic side effects, and the development of novel agents is still underway.
The secreted protein, short palate lung and nasal epithelial clone 1 (SPLUNC1) is abundant within the ASL. In normal airways SPLUNC1 regulates ASL volume by binding to and preventing the cleavage of ENaC, thereby inhibiting its action.10 Importantly, SPLUNC1 becomes ineffective at inhibiting ENaC when the ASL pH is reduced. In the acidic, dehydrated CF airway, normal ENaC regulation is disrupted, resulting in sodium hyper-absorption and further depletion of ASL volume.14 Protease inhibitors and pH-insensitive derivatives of SPLUNC1 could prove beneficial in regulating ENaC activity, but further studies of these targets in human models are required.
ENaC regulation rather than inhibition may be required to maintain normal ASL homeostasis. A large-scale siRNA screen was performed to identify genes involved in the mechanistic processes for ENaC regulation. This was coupled with live cell microscopy in human airway epithelial cells to assess ENaC function.15 Over 1500 activators and inhibitors for ENaC were identified and further detailed investigation highlighted a particular protein, DGKι, as a potential target. DGKι is required to maintain ENaC activity, and its inhibition reduced ENaC activity in CF primary airway epithelial cells. Furthermore, ENaC activity and airway fluid absorption were normalised, highlighting its potential role as a novel CF therapy.15
Alternative chloride channels and transporters
Although ENaC regulation will benefit sodium hyper-absorption, the correction of dysfunctional chloride and bicarbonate transport will require additional strategies potentially involving the modification of other alternative channels (figure 1).16
One such approach is to target calcium activated chloride channels (CaCC). These are widely expressed in a range of tissue types and are diverse in their functions. Anoctamin 1 (Ano1) is a member of the anoctamin transmembrane protein family and is an essential component of the CaCC.17 Amongst many other tissues, it is expressed in the airway epithelium and is active in CF airways. Ano1 knockout mice develop a CF-like phenotype, highlighting its importance in chloride transport and potential role for therapy.18 Upregulation of Ano1 may be successful in compensating for CFTR dysfunction in CF.
Challenges have been faced with developing agents that utilise CaCC pathways, initially due to poor airway potency. An alternative longer-acting P2Y2 receptor agonist, denufosol, did not show any significant improvements in lung function in phase III trials. Although this area holds promise, further investigation is required to establish potential therapies.
Another approach would be to target the solute carriers (SLC), a family of plasma membrane anion exchangers and channels that are widely expressed throughout the body. In particular, the SLC26A members of this family are involved in chloride/bicarbonate exchange in secretory and airway epithelia and have been found to have regulatory interactions with CFTR.19 ,20 A greater understanding of these interactions together with further investigation of their significance in the CF airway could potentially provide some promising potential targets for therapy.
Conclusions and future directions
The ASL plays a key role in mucus clearance and lung defence. In addition to CFTR, additional apical channels and transporters are involved in ion transport, and could be potential strategies for CF therapy.
Challenges may be faced given the wide expression of many of these proteins, the subsequent risk of potential unwanted systemic effects and the additional challenge of achieving adequate drug potency within the airways. Furthermore, the dynamic assessment of the ASL in vivo is challenging, as is the functional assessment of its response to future druggable targets.
Further development of representative human models would be necessary to enable the investigation of these promising avenues for future CF management. Targeting a combination of these bypass channels could successfully correct the ASL abnormalities seen in CF, providing benefit to all patients regardless of their mutation.
References
Footnotes
Contributors All authors developed a plan for the article, IJH and MB wrote the first draft that all authors commented on, IJH and MB then completed the final version.
Funding MAG, CW and MB: Strategic Research Centre Grant (SRC003), Cystic Fibrosis Trust. MB: Medical Research Council Clinician Scientist Fellowship (MR/M008797/1). JPG: Newcastle University Wellcome Trust Institutional Strategic Support Fund.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.