Article Text

Abstract
Introduction Increasing evidence links COPD pathogenesis with pulmonary capillary apoptosis. We previously demonstrated that plasma levels of circulating microparticles released from endothelial cells (EMPs) due to apoptosis are elevated in smokers with normal spirometry but low diffusion capacity, that is, with early evidence of lung destruction. We hypothesised that pulmonary capillary apoptosis persists with the development of COPD and assessed its reversibility in healthy smokers and COPD smokers following smoking cessation.
Methods Pulmonary function and high-resolution CT (HRCT) were assessed in 28 non-smokers, 61 healthy smokers and 49 COPD smokers; 17 healthy smokers and 18 COPD smokers quit smoking for 12 months following the baseline visit. Total EMP (CD42b−CD31+), pulmonary capillary EMP (CD42b−CD31+ACE+) and apoptotic EMP (CD42b−CD62E+/CD42b−CD31+) levels were quantified by flow cytometry.
Results Compared with non-smokers, healthy smokers and COPD smokers had elevated levels of circulating EMPs due to active pulmonary capillary endothelial apoptosis. Levels remained elevated over 12 months in healthy smokers and COPD smokers who continued smoking, but returned to non-smoker levels in healthy smokers who quit. In contrast, levels remained significantly abnormal in COPD smokers who quit.
Conclusions Pulmonary capillary apoptosis is reversible in healthy smokers who quit, but continues to play a role in COPD pathogenesis in smokers who progressed to airflow obstruction despite smoking cessation.
Trial registration number NCT00974064; NCT01776398.
- Smoking cessation
- COPD ÀÜ Mechanisms
- Tobacco and the lung
Statistics from Altmetric.com
Key messages
What is the key question?
Is pulmonary capillary apoptosis, as measured by plasma levels of microparticles released from apoptotic endothelial cells (EMPs), reversible in healthy smokers and COPD smokers following smoking cessation?
What is the bottom line?
Pulmonary capillary apoptosis is reversible in healthy smokers who quit, but not in COPD smokers despite 12 months of smoking cessation, suggesting that the apoptosis continues to play a role in COPD pathogenesis in smokers who progressed to airflow obstruction despite smoking cessation.
Why read on?
Our longitudinal study demonstrates persistent endothelial stress in subjects with COPD despite smoking cessation and provides a biological correlate to the epidemiological data showing that smoking cessation only has a moderate effect on the continuous decline of lung function in COPD smokers, suggesting EMP levels might serve as a useful biomarker to follow smoking-associated endothelial apoptosis.
Introduction
COPD, the third leading cause of mortality in the USA, is defined by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as a chronic lung disorder with airflow limitation that is not fully reversible.1 There is overwhelming evidence that most cases of COPD are caused by cigarette smoking, with approximately 20% of smokers at risk for COPD if they continue to smoke.2 The airflow obstruction that characterises COPD is caused by a variable mixture of small airway disease (bronchitis) and parenchymal destruction (emphysema).1 ,3 Although the airway and alveolar diseases were classically considered as separate entities, it is now recognised that they usually coexist to variable degrees and are closely linked, with the parenchymal destruction evolving around areas of small airway disease.3–7
There is increasing evidence that the pathogenesis of COPD is linked, in part, to apoptosis of pulmonary capillaries.8–11 Consistent with this concept, we recently demonstrated that smokers and, to a greater extent, smokers with early evidence of lung destruction (normal spirometry, but low diffusing capacity (DLCO)) have elevated levels of circulating endothelial microparticles (EMPs).12 Importantly, a significant proportion of these EMPs are derived from pulmonary capillaries and have characteristics of apoptotic EMPs, that is, they are derived from lung endothelial cells that have been induced to undergo apoptosis.12–14
If elevated levels of circulating apoptotic EMPs are a reflection of active smoking-related injury, to lung endothelium, based on the knowledge that even those COPD smokers who stop smoking continue to have a decline in lung function that is more rapid than that of healthy non-smokers or healthy smokers who quit smoking,15 we hypothesised that elevated levels of circulating apoptotic EMPs may persist in COPD smokers following smoking cessation, reflecting continuous lung endothelial injury that persists even after the stress of smoking is removed. To assess this hypothesis, we quantified the levels of circulating EMPs, and the fraction represented by apoptotic EMPs, in non-smokers, healthy smokers and smokers with COPD at baseline and at three more intervals over 1 year and then compared those levels with those obtained from a subgroup of healthy smokers and COPD smokers who successfully stopped smoking after baseline assessment. The data demonstrate that circulating EMP levels derived from apoptotic pulmonary capillary endothelial cells remain elevated over 1 year in healthy smokers and COPD smokers who continue smoking. However, while levels of total and apoptotic EMPs return to non-smoker levels in healthy smokers who successfully quit smoking, total and apoptotic EMP levels remain elevated in COPD smokers who quit smoking persisting 12 months of smoking cessation.
Methods
Human subjects and clinical phenotypes
All subjects were evaluated at the Weill Cornell NIH Clinical and Translational Science Center and Department of Genetic Medicine Clinical Research Facility, under the clinical protocols approved by the Institutional Review Board. Recruitment was from the general population in New York City by posting advertisements in local newspapers and on electronic bulletin boards. All subjects provided written consent prior to enrolment and then underwent thorough medical history, screening and pulmonary function tests. Smoking status was determined based on self-reported history and quantified levels of urine nicotine metabolites. For details and full inclusion/exclusion criteria, see online supplementary methods. A total of 138 subjects were assessed for circulating total and apoptotic EMP levels at baseline, 3, 6 and 12 months (28 non-smokers, 61 healthy smokers and 49 COPD GOLD I/II smokers). See online supplementary figure S1 for study design.
Supplemental material
Characterisation of plasma EMPs
EMPs were quantified according to a standard operating procedure as previously described12 to eliminate variability in sample processing. Briefly, blood was collected, processed within 1 hour and stained for the endothelial markers PECAM (CD31) and E-selectin (CD62E) and the constitutive platelet-specific glycoprotein Ib (CD42b) to differentiate endothelium-originated microparticles from platelet-derived microparticles, which also express CD31. EMPs were defined as microparticles <1.5 μm in size, expressing CD31+ or CD62E+ but not CD42b. We have previously shown that staining with annexin V is comparable with CD42b−CD31+ staining,12 but annexin V was not used because it is not specific for EMPs.16 Circulating EMPs are present in low levels in plasma of healthy subjects, reflecting normal endothelial turnover,17 but their levels increase in a variety of vascular-related disorders. As in our previous study,12 total EMP levels above the non-smoker total EMP mean level plus 2 SDs were considered abnormally elevated. To assess the presence of relative contribution of pulmonary capillary endothelium to the elevated total EMP levels,1 ,12 EMPs were co-stained with antihuman ACE inhibitors, which is abundantly expressed on pulmonary capillary endothelium (CD42b−CD31+ACE+).13 To quantify the proportion of EMPs that originated from apoptotic endothelium, we assessed the ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in all groups. EMPs induced by apoptosis express the constitutive CD31 marker, whereas activation-induced EMPs express CD62E. Using these criteria, EMPs with a low CD42b−CD62E+ to CD42b−CD31+ ratio were defined as ‘apoptotic EMPs’, and the percentage of subjects with apoptotic EMPs with CD42b−CD62E+/CD42b−CD31+ ratio below the lowest ratio in healthy non-smokers was quantified. See online supplementary methods for further details on the EMP analysis.
Assessment after smoking cessation
After the baseline levels of total and apoptotic EMPs were determined, all healthy smokers and COPD smokers were invited to stop smoking using a combination of varenicline and counselling for 3 months (see details in online supplementary methods). A total of 17 healthy smokers and 18 COPD smokers successfully quit smoking as confirmed by urine tobacco metabolite level quantification at 3, 6 and 12 months after the baseline; subjects were considered true quitters only if there were no detectable levels of nicotine metabolites in the urine at months 3, 6 and 12. Healthy smokers and smokers with COPD were treated with exact same prescription of varenicline to prevent any effect it might have on EMP levels. All other healthy smokers and COPD smokers were considered current smokers if urine cotinine level was ≥104 ng/mL at each time point, a level based on our previous study of low-level smoke exposure,18 where 104 ng/mL was calculated as the induction half-maximal level (ID50) at which the small airway epithelium, the initial site of smoking-related pathology, showed an abnormal response. See online supplementary figure S1 for study design.
Statistical analysis
χ2 test, with a Yates' correction for small sample size, was used for comparing demographic parameters and the number of subjects with high total EMP and apoptotic EMP levels, and pairwise analysis of variance was used to compare total and apoptotic EMP levels between groups and within a group, at different time points with no correction for multiple test, as the number of tests was low (<21). In order to eliminate the effect of diseases known to be associated with elevated EMPs, including diabetes19 ,20 and systemic hypertension,21 or drugs for COPD, including corticosteroids and bronchodilators, subjects with known disease state or drug treatment were removed from statistical analysis. Removal of those subjects did not alter the results.
Results
Study population
Except for minor differences, the study population of non-smokers was comparable with the healthy smokers and COPD smokers in all demographic parameters (see table 1 for details). At each time point, the non-smokers had undetectable urine nicotine (not shown) and cotinine levels (figure 1). Both the healthy smokers and COPD smokers who continued smoking had urine cotinine levels consistent with tobacco smoking and comparable in both groups at each time point (p>0.07, all comparisons). In healthy smokers and COPD smokers who quit smoking, urine nicotine and cotinine levels were consistent with smoking at baseline and were undetectable at all time points after baseline (see online supplementary methods for details of urine nicotine metabolite level criteria for smoking/abstinence).
Study Population*
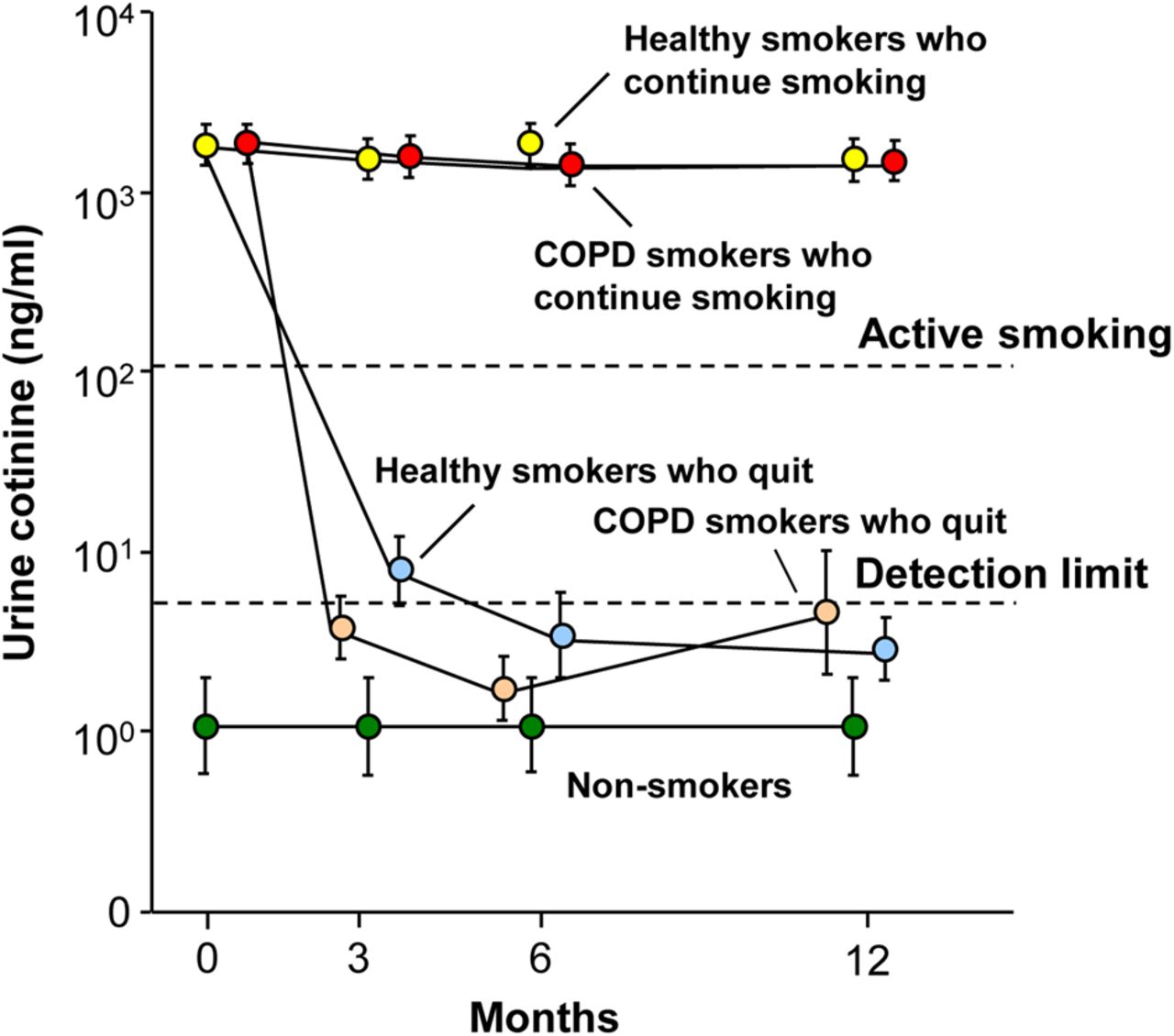
Urine cotinine levels (ng/mL) as a measure of smoking status at baseline and at 3, 6 and 12 months in non-smokers, healthy smokers and smokers with COPD (COPD smokers). Shown are data for non-smokers (n=28, green circles), healthy smokers who continue to smoke (n=44, yellow circles), healthy smokers who quit smoking following baseline (n=17, light blue circles), COPD smokers who continue to smoke (n=31, red circles) and COPD smokers who quit smoking following baseline (n=18, tan circles). Data represent mean±SE. Dashed lines indicate urine cotinine detection level of ≤5 ng/mL and urine cotinine level of ≥104 ng/mL for active smoking (see online supplementary methods). EMP, endothelial microparticles.
Total EMP levels
Consistent with our prior study with a different cohort,12 total EMP levels were higher in healthy smokers compared with non-smokers (figure 2A, p<0.005). In addition, COPD smokers had elevated levels of total EMPs compared with non-smokers (p<0.007), but lower than those of healthy smokers (p<0.02). Twenty-two (36%) healthy smokers and eight (16%) COPD smokers had high levels of total EMPs (p<0.03). There was no correlation between the level of total EMPs and any pulmonary function or demographic parameters (r2<0.08, all correlations, see online supplementary figure S2). For the COPD group, total circulating EMP levels were independent of drugs used for treatment, including inhaled and systemic corticosteroids and bronchodilators.

Levels of total circulating endothelium microparticles (EMPs), ACE+ EMPs and apoptotic EMPs in plasma at baseline. Shown are data for non-smokers (n=28, green circles), healthy smokers (n=61, yellow circles) and smokers with COPD (n=49, red circles). (A) Total CD42b−CD31+ EMPs. The grey shaded area indicates the non-smoker mean±2 SDs. The % value above the smoker populations represents the proportion of smokers with EMP levels above that mean. (B) CD42b−CD31+ACE+ EMPs. Proportion of total CD42b−CD31+ EMPs in plasma that express ACE+. The grey shaded area represents the non-smoker mean±2 SDs. (C) Ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs. The dashed line indicates the lowest ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in non-smokers. The % value below the smoker populations represents the proportion of smokers with a ratio below that level. (A–C) Bold dashed lines represent the mean for each group. Symbols inside the dots: A horizontal line indicates subjects with systemic hypertension; a vertical line indicates subjects with type 2 diabetes mellitus; a star indicates subjects with type 1 diabetes.
Origin of the circulating EMPs
In our prior study of circulating EMPs,12 we demonstrated that most of the circulating CD42b−CD31+ EMPs were positive for ACE inhibitors , a surface protein more highly expressed on pulmonary capillary endothelium than in other endothelial beds.13 In the present study, an average of 75% of the circulating EMPs in all subjects were CD42b−CD31+ACE+. There were similar levels of ACE+ EMPs in the healthy smoker group compared with the non-smokers or COPD smokers (figure 2B, p>0.1, both comparisons), and higher levels of ACE+ EMPs in the COPD smoker group compared with the non-smokers (p<0.0001).
EMPs derived from apoptotic endothelium
To quantify the proportion of EMPs originating from apoptotic endothelium, we assessed the ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in all groups. The CD42b−CD62E+/CD42b−CD31+ EMP ratio in non-smokers was distributed around a mean of 0.9, significantly higher than that in healthy smokers (mean 0.6; a lower ratio indicates greater number of apoptotic EMPs) and COPD smokers (mean 0.55, p<0.0001, both groups compared with non-smokers; p>0.6, COPD smokers compared with healthy smokers). CD42b−CD62E+/CD42b−CD31+ EMPs below the lowest level in non-smokers were defined as apoptotic EMPs with 48% of healthy smokers and 45% of COPD smokers having increased levels of apoptotic EMPs (p>0.7), that is, even though there are less subjects with total circulating EMPs in COPD smokers compared with healthy smokers (figure 2A), the relative proportion of subjects with apoptotic EMPs was similar (figure 2C), implying that there is active pulmonary capillary apoptosis ongoing in both the healthy smokers and COPD smokers. There was no correlation of CD42b−CD62E+/CD42b−CD31+ EMP ratio with any lung function or demographic parameter (r2=0.09, all correlations, see online supplementary figure S3). Within the COPD smoker group, there was no correlation of total CD42b−CD31+ EMP levels or CD42b−CD62E+/CD42b−CD31+ EMP ratio to the DLCO (% predicted) or % emphysema on high-resolution CT (HRCT) (r2=0.04, all comparisons; not shown), suggesting these parameters are likely measuring different aspects of the destruction process.
Effect of smoking cessation on total EMP levels
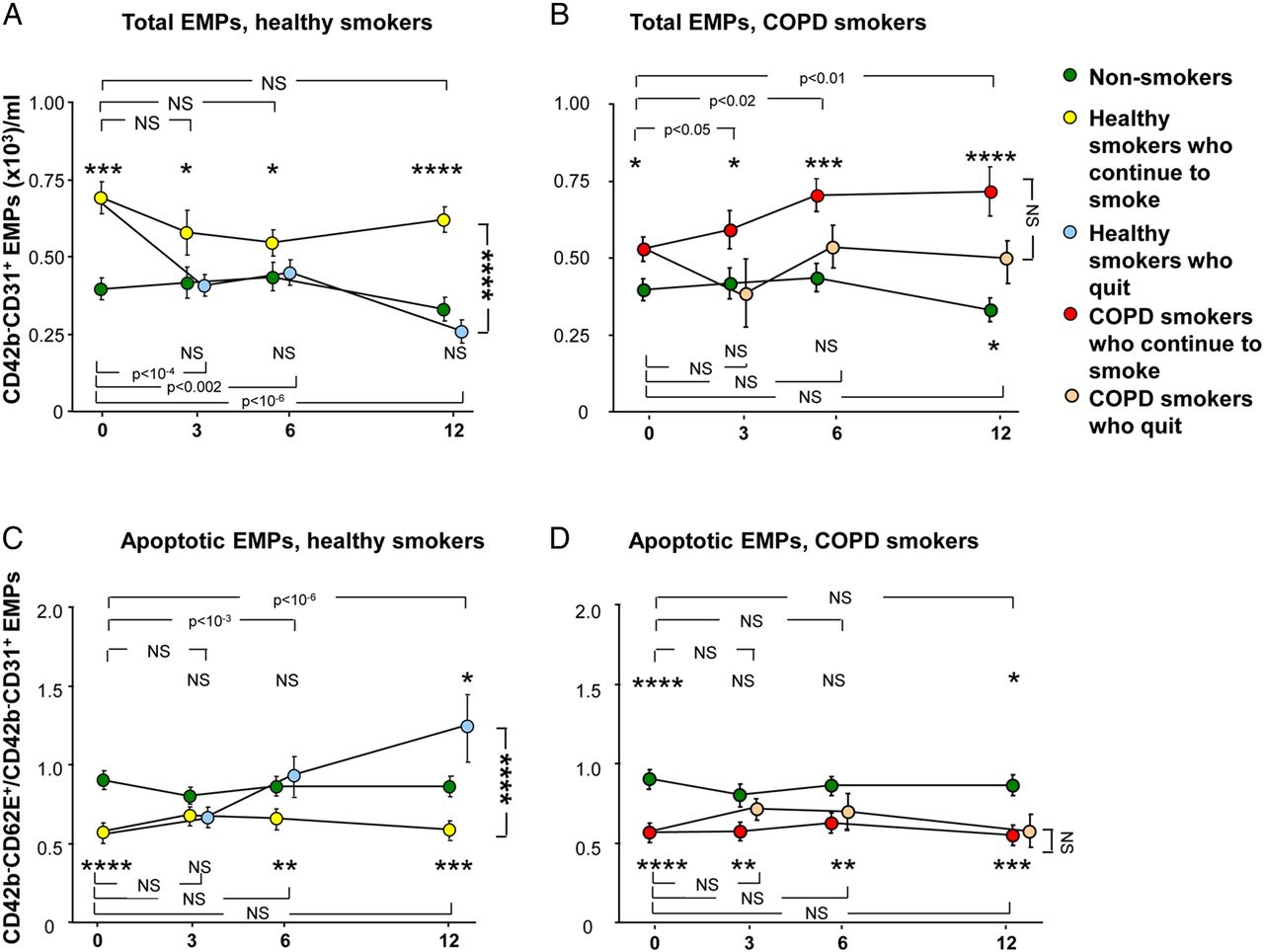
The levels of total EMPs were followed for a period of 1 year at baseline, 3, 6 and 12 months in non-smokers, healthy smokers and COPD smokers (figure 3A, B). In non-smokers, total EMP levels were stably low throughout the duration of the study (p>0.6, each time point compared with baseline). In healthy smokers who continued smoking, the levels were stably high at each time point (p>0.1, each time point compared with baseline; p<0.05, each time point compared with the non-smokers at the same time point). In contrast, in the healthy smokers who quit smoking, total EMP levels significantly decreased following smoking cessation to the levels of non-smokers (p<0.002, each time point compared with baseline; p>0.4, compared with non-smokers at the same time point at 3, 6 and 12 months, figure 3A). At 12 months, the total EMP levels in healthy smokers who continued smoking were significantly higher compared with healthy smokers who quit (p<10−3). Total EMP levels in COPD smokers who continued smoking were stably elevated compared with non-smokers at each time point (p<0.05, all comparisons). In contrast to the healthy smokers, in COPD smokers who quit smoking, total EMP levels initially decreased following smoking cessation at month 3, but became elevated again at 6 and 12 months (figure 3B). The levels were not significantly different compared with non-smokers at 3 and 6 months (p>0.1, both comparisons), but were significantly elevated at 12 months (p<0.05) and similar to those of COPD smokers who continued smoking (p>0.08).

{kind=link}
{kind=link}
{kind=link}
Total circulating CD42b−CD31+ endothelial microparticles (EMPs) and ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs over time in non-smokers (n=28, green circles), healthy smokers who continue to smoke (n=44, yellow circles), healthy smokers who quit smoking following baseline (n=17, light blue circles), smokers with COPD (COPD smokers) who continue smoking (n=31, red circles) and COPD smokers who quit smoking following baseline (n=18, tan circles). (A and B) Total CD42b−CD31+ EMPs. (C and D) Ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs. (A and C). Healthy smokers who continue to smoke and healthy smokers who quit smoking versus non-smokers. (B and D) COPD smokers who continue to smoke and COPD smokers who quit smoking versus non-smokers. (A–D) Data represent mean±SE. p Values comparing each time point to baseline within the same group are shown at the top of the panel (for the group, ie, above the non-smokers at month 12) and at the bottom of the panel (for the group, ie, below the non-smokers at month 12). p Values comparing each time point in a smoker group to the same time point in the non-smoker group are shown above the group, if the group is above the non-smokers at month 12 and below the group if the group is below the non-smokers at month 12. p Values comparing the subjects who continue to smoke with those who quit smoking at month 12 are to the right of the panel. NS, not significant; *, **, ***, **** indicate p<0.05, p<0.01, p<0.001 and p<0.0001, respectively.
Effect of smoking cessation on apoptotic EMP levels
The ratio of CD42b−CD62E+/CD42b−CD31+ EMPs was stably high in non-smokers and stably low (ie, EMPs were apoptotic-derived) in healthy smokers who continued smoking (p>0.1, each time point compared with baseline within the non-smoker group and within the healthy smoker group; p<0.01, healthy smokers who continued smoking compared with non-smokers, at baseline, months 6 and 12; figure 3C). Interestingly, in the healthy smokers who quit smoking group, the ratio increased following smoking cessation (p<10−3, within the healthy smokers who quit group, at months 3 and 6 compared with baseline, a p value significant even with correction for multiple tests) to the level of non-smokers at months 3 and 6 (p>0.1, both comparisons compared with non-smokers at the same time point) and superseded that of non-smokers at month 12 (p<0.05). The ratio was significantly higher (ie, less apoptotic-derived EMPs) in healthy smokers who quit smoking compared with smokers who continued smoking at month 12 (p<10−3, a p value significant even with correction for multiple tests). There were no significant changes in the CD42b−CD62E+/CD42b−CD31+ ratio in COPD smokers who continued smoking (p>0.1, within the COPD smoker group, each time point compared with baseline), and it remained significantly low compared with non-smokers at each time point (p<0.01, all comparisons, figure 3D). In contrast to the healthy smoker group, in COPD smokers who quit smoking, there was no change in the ratio (p>0.1, within the COPD who quit group, each time point compared with baseline), and the ratio remained significantly low compared with non-smokers at baseline and month 12 (p<0.05, both comparisons). The ratio at month 12 was similarly low (ie, more apoptotic-derived EMPs) in COPD smokers who continued smoking and in COPD smokers who quit (p>0.4).
Discussion
COPD is a chronic, debilitating disease that is caused primarily by cigarette smoking.1 ,3–5 Cigarette smoke is very complex, with 1014 oxidants and >4000 compounds stressing the lung with each puff.22 The apoptotic loss of pulmonary capillaries in association with smoking is well recognised,8–10 although it is not known whether this represents the primary mechanism of lung destruction associated with smoking, a subtype of lung destruction, or is secondary to other mechanisms, such as inflammatory cell-mediated processes.5 ,6 ,9 ,10 ,23 Using circulating, total and apoptotic-endothelial cell microparticles as biomarkers for pulmonary capillary apoptosis, the data in the present study document that pulmonary capillary endothelial apoptosis is a persistent process in smokers with and without COPD. In healthy smokers who quit smoking, the levels of total and apoptotic EMPs return to the levels of non-smokers over time. In contrast, in COPD smokers who quit smoking, the levels of total and apoptotic EMPs remain abnormal over 1 year and were still significantly different compared with non-smoker levels at 12 months. The majority of the COPD subjects assessed for EMPs in this study were GOLD I and GOLD II, providing evidence for ongoing pulmonary endothelial apoptosis even in the earliest stages of COPD. Importantly, the FEV1/FVC ratio of the COPD smokers was 0.63±0.06, on average, well below the 0.7 ratio threshold definition of COPD GOLD I.
These observations were not altered by the removal of subjects with diseases known to be associated with elevated EMPs, suggesting that smoking has a much stronger effect on EMP levels than hypertension or diabetes.
Endothelial microparticles
Different cell types respond to cell activation, injury and/or apoptosis by shedding submicron membrane vesicles, called microparticles, from their plasma membranes.16 ,24 Microparticles detected in plasma are of various cellular origins, predominantly derived from platelets, leucocytes and endothelial cells.24 Endothelial microparticles, defined as CD42b−CD31+ or CD42b−CD62E+ microparticles, can be generally distinguished from microparticles of other cell types by their size (0.1–1.5 µm), constitutive expression of the platelet–endothelial cell adhesion marker CD31 (PECAM) and the absence of the platelet-specific glycoprotein Ib marker CD42b.24 ,25 Apoptosis-induced EMPs express CD31, whereas activation-induced EMPs express CD62E. In this regard, a low ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs can be used as an index of apoptosis.24 ,25 EMPs variably co-express phosphatidylserine (annexin V).25 ,26
EMPs can be found in the plasma of healthy subjects;24 however, increased levels are associated with vascular disease and endothelial dysfunction in atherosclerosis and acute coronary syndrome,24 ,27 acute ischaemic stroke,24 ,28 end-stage renal failure,29 pre-eclampsia and gestational hypertension,30 hypertension,21 ,24 ,29 pulmonary hypertension,31 metabolic syndrome,32 venous thromboembolism33 and obstructive sleep apnoea.34 Consistent with our prior study12 and the data in the present study, Heiss and colleagues35 have shown healthy non-smokers exposed for 30 min to low levels of cigarette smoke had increased circulating EMP levels.
Clinical measures of alveolar capillary destruction
The observation of endothelial apoptosis in the lungs of humans with emphysema is well documented. There is increased DNA fragmentation in the pulmonary capillaries and arteriolar endothelium of subjects with COPD, and increased alveolar endothelial and epithelial cell death in human emphysematous lungs compared with lungs of non-smokers or smokers without emphysema.9 ,10 Lung levels of alveolar epithelial-derived vascular endothelial growth factor are decreased in emphysema, contributing to the complex mechanisms of pulmonary capillary endothelial destruction.10
The data in the present study add total and apoptotic EMP levels to a growing list of biomarkers that may be useful in assessing active destruction and defining subclinical molecular phenotypes of lung disease.17 ,36 ,37 As described in the editorial by Chandra and colleagues,14 accompanying our study of apoptotic EMPs in healthy smokers and smokers with normal spirometry but low DLCO, the observation of elevated levels of apoptotic EMPs may represent early lung destruction and a subphenotype of subjects with lung destruction. The observation in the present study that, on average, a fraction of smokers with COPD also have elevated levels of total and apoptotic EMPs is consistent with the discussion of the concept of vascular subphenotypes of COPD by Chandra et al.14 In this context, the data on circulating and apoptotic EMPs support the idea that, while the global concept of COPD as an FEV1-defined disorder is useful for epidemiologic studies and as a paradigm for routine clinical care, it is likely masking the concept that there are several subphenotypes of COPD.38–42 Consistent with this concept, correlation of the total EMP levels and apoptotic EMP levels with the conventional measures of lung destruction (DLCO and HRCT) was, at best, very weak. This may imply that, while overlapping, each parameter is measuring a somewhat different aspect of the same process and/or that each parameter is assessing a different subpopulation of what is globally referred to as ‘lung destruction’.
Our longitudinal study demonstrates that total and apoptotic EMP levels remain stable over a period of 1 year in subjects with no change to their smoking habits. Intervention with smoking cessation can normalise the levels of total and apoptotic EMPs in healthy smokers, but in contrast, cessation did not lead to significant changes in total and apoptotic EMP levels in COPD smokers in our study. This observation provides a biological correlate for epidemiological data showing that smoking cessation only has a moderate effect to slow the decline of lung function in COPD smokers, that is, we believe that these data show that EMP levels might serve as a useful biomarker to follow smoking-associated endothelial apoptosis. The longitudinal aspect of this study demonstrates persistent endothelial stress in subjects with COPD, despite smoking cessation, and may help to explain the irreversible lung destruction associated with most cases of COPD, as evidenced by lung function of COPD smokers following smoking cessation that does not return to normal,15 and may serve as a basis for additional studies of the mechanisms of the continuous pulmonary damage leading to this persistent EMP release.
Acknowledgments
The authors thank M Elnashar, A Rogalski, E Blass, S Mootoo and T Wilson for their help in processing blood samples; the Clinical Operations and Regulatory Affairs, Department of Genetic Medicine for help with these studies; and N Mohamed for help in preparing this manuscript. These studies were supported, in part, by P50HL084936, R01HL107882, U01HL121828, UL1 TR000457, UL1 RR02414 and Hoffmann-La Roche.
References
Footnotes
Contributors Conception and design: RGC. Acquisition and data interpretation: YS-B, MRS, AK, CG, AET, BG-H, RJK, CH, JGM, HB, SS, SGP, HH, GW, CSS, SV, JSF and RGC. Drafting of the manuscript: YS-B, MRS, AK, AET and RGC.
Funding F. Hoffman-La Roche, National Institutes of Health (P50HL084936), (R01HL107882), (U01HL121828) (UL1 RR02414) and (UL1 TR000457).
Competing interests Hans Bitter, Sriram Sridhar, Sreekumar G. Pillai, Holly Hilton, Gerhard Wolff, Christopher S. Stevenson, Sudha Visvanathan and Jay S. Fine were all former employees of Hoffmann-La Roche.
Ethics approval Weill Cornell Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.