Article Text

Abstract
Rationale Cigarette smoke exposure is associated with an increased risk of the acute respiratory distress syndrome (ARDS); however, the mechanisms underlying this relationship remain largely unknown.
Objective To assess pathways of lung injury and inflammation in smokers and non-smokers with and without lipopolysaccharide (LPS) inhalation using established biomarkers.
Methods We measured plasma and bronchoalveolar lavage (BAL) biomarkers of inflammation and lung injury in smokers and non-smokers in two distinct cohorts of healthy volunteers, one unstimulated (n=20) and one undergoing 50 μg LPS inhalation (n=30).
Measurements and main results After LPS inhalation, cigarette smokers had increased alveolar-capillary membrane permeability as measured by BAL total protein, compared with non-smokers (median 274 vs 208 μg/mL, p=0.04). Smokers had exaggerated inflammation compared with non-smokers, with increased BAL interleukin-1β (p=0.002), neutrophils (p=0.02), plasma interleukin-8 (p=0.003), and plasma matrix metalloproteinase-8 (p=0.006). Alveolar epithelial injury after LPS was more severe in smokers than non-smokers, with increased plasma (p=0.04) and decreased BAL (p=0.02) surfactant protein D. Finally, smokers had decreased BAL vascular endothelial growth factor (VEGF) (p<0.0001) with increased soluble VEGF receptor-1 (p=0.0001).
Conclusions Cigarette smoke exposure may predispose to ARDS through an abnormal response to a ‘second hit,’ with increased alveolar-capillary membrane permeability, exaggerated inflammation, increased epithelial injury and endothelial dysfunction. LPS inhalation may serve as a useful experimental model for evaluation of the acute pulmonary effects of existing and new tobacco products.
- ARDS
- Tobacco and the lung
Statistics from Altmetric.com
Key messages
What is the key question?
How does cigarette smoke exposure predispose patients to develop acute respiratory distress syndrome (ARDS)?
What is the bottom line?
After lipopolysaccharide exposure, smokers have exaggerated alveolar-capillary barrier permeability, inflammation and epithelial injury, suggesting that priming of these pathways may contribute to the increased risk of ARDS found in smokers.
Why read on?
To our knowledge, this study is the first to use a human experimental model to focus on directly investigating the mechanisms through which cigarette smoke exposure predisposes patients to develop ARDS.
Introduction
Although many harmful effects of tobacco have been known for decades, cigarette smoke exposure has only recently been identified as a risk factor for acute respiratory distress syndrome (ARDS). Over the past several years, studies in a variety of populations have found an increased risk of ARDS among smokers. In critically ill patients with blunt trauma, active and passive cigarette smoke exposure have been associated with an increased risk of ARDS;1 likewise, smoking is associated with an increased risk of ARDS in non-pulmonary sepsis2 and following blood transfusion,3 as well as with primary graft dysfunction,4 a form of ARDS that occurs within 72 h of lung transplant. Furthermore, cigarette smokers who develop ARDS do so with fewer comorbidities and at a younger age,5 suggesting that smokers may be more prone to developing ARDS when they are less severely ill. However, the mechanisms underlying the relationship between cigarette smoke and ARDS remain poorly characterised.
Potential mechanisms by which cigarette smoking may predispose patients to develop ARDS have largely been extrapolated from other experimental settings. Cigarette smoke has numerous acute effects on the lung, several of which are implicated in ARDS pathogenesis, including alveolar inflammation,6 increased alveolar epithelial permeability,7 increased pulmonary endothelial permeability8 and platelet dysfunction.9 In a recent study of ex vivo human lungs rejected for organ transplantation, donor smoking was associated with increased pulmonary oedema, with evidence of inflammation and epithelial injury in bronchoalveolar lavage (BAL), whereas heavy smoking was associated with impaired alveolar fluid clearance.10 Another study found that cigarette smokers with ARDS had higher oedema fluid to plasma protein ratios, reflecting increased alveolar-capillary barrier permeability.11 Additional human clinical studies with direct relevance to ARDS are needed to obtain a better understanding of the mechanisms through which cigarette smoke predisposes patients to ARDS, with the ultimate goal of developing new preventative and therapeutic strategies for this frequently fatal syndrome as well as identifying biomarkers that might become the basis for regulation of existing and new tobacco products.
In this study, we compared the association of cigarette smoke exposure with plasma and BAL biomarkers of lung injury and inflammation, chosen a priori, in two groups to (1) test if cigarette smoke increases alveolar-capillary membrane permeability in response to a ‘second hit’, and (2) study mechanistic biomarkers that might help to explain differences in alveolar-capillary permeability between smokers and non-smokers. We hypothesised that cigarette smoke exposure primes the lung to develop increased alveolar-capillary membrane permeability, and ultimately ARDS, through an exaggerated inflammatory and injurious response to a ‘second-hit,’ which we modelled with inhaled lipopolysaccharide (LPS). Some of the results of this study have been previously reported in the form of an abstract.
Subjects and methods
Subjects
Samples from two previously enrolled cohorts were analysed in this study. The first cohort comprised healthy outpatients enrolled in an elective bronchoscopy study at the University of Colorado.6 Subjects, who had no history of cardiac, lung, liver or renal dysfunction, underwent bronchoscopy with BAL. Details of the bronchoscopy are available in the online supplementary data. We used BAL samples from 20 healthy subjects without a history of alcohol use disorders. Of these 20 subjects, 10 were active smokers by self-report. The study was approved by the University of Colorado institutional review board. All subjects provided consent for participation, including for the use of samples in future studies.
The second cohort consisted of healthy non-alcoholic volunteers enrolled in a study at Queen's University, Belfast, UK.12 This study was originally designed to assess the anti-inflammatory effects of simvastatin in humans exposed to LPS. Of the 30 enrolled subjects, nine were active smokers by self-report. Subjects were randomised to either simvastatin or placebo for 4 days and then exposed to 50 μg inhaled LPS (Escherichia coli serotype O26:B6; Sigma Chemicals, Poole, Dorset, UK). Bronchoscopy with BAL was performed 6 h after LPS inhalation. Details of the LPS inhalation and bronchoscopy are available in the online supplementary data. Plasma was obtained both before LPS administration and 24 h afterwards. After the conclusion of this study, four additional smokers were enrolled at Queen's University, Belfast; although recruitment was similar to the initial 30 Belfast subjects, these four subjects did not receive statins or placebo. Additionally, these four subjects underwent two bronchoscopies—first, without LPS stimulation (with plasma drawn before the procedure and 24 h later), and then 4 weeks later, LPS inhalation followed by bronchoscopy with BAL 6 h later (with plasma drawn according to the previous studies). The study was approved by the local research ethics committee. All subjects provided consent for participation, including for the use of samples in future studies.
Measurements
Total protein
Total protein was measured in BAL samples only by the Bradford assay (Bio-Rad Laboratories, Hercules, California, USA).
Inflammatory biomarkers
Interleukin (IL)-8 and IL-1β were measured in plasma and BAL by ELISA (R&D Systems, Minneapolis, Minnesota, USA). Matrix metalloproteinases (MMP)-1, -2, -3, -7, -8 and -9 were measured in LPS stimulated plasma by cytometric bead array (R&D systems). Only MMP-8 was measured in baseline plasma (ELISA, R&D Systems). Polymorphonuclear neutrophils (PMNs) were measured in BAL only by manual count.
Surfactant protein D
Surfactant protein D (SP-D) was measured in plasma and BAL using ELISA (Yamasa Corporation, Tokyo, Japan).
Vascular endothelial growth factor and receptor
Vascular endothelial growth factor (VEGF) was measured in plasma and BAL using ELISA (R&D systems). Soluble VEGF receptor-1 (sVEGFr-1) was measured in BAL alone using ELISA (R&D systems).
Statistical analysis
Normally distributed variables were compared using a Student's t test and displayed as mean±SD. Non-normally distributed variables were compared using the Mann–Whitney U test and displayed as median with IQR. In the Belfast cohort, linear regression was performed to assess whether the differences in biomarkers between smokers and non-smokers were independent of statin exposure. Regression with an interaction term was used to compare the mean difference in each biomarker between smokers and non-smokers at baseline and after LPS inhalation; this analysis formally tests the hypothesis that the association between LPS administration and the biomarker of interest differs in smokers as compared with non-smokers. Interaction was adjusted for age as a possible confounder. Log transformation was used, as needed, to fulfil all assumptions required for linear regression testing. A p value ≤0.05 was considered statistically significant. Statistical analyses were performed with STATA V.13.1 (StataCorp LP, College Station, Texas, USA).
Results
Demographics
All subjects were healthy without comorbidities. There was no difference in sex distribution between cohorts. The Colorado cohort was older than the Belfast cohort (mean age 40 vs 26 years, p<0.0001). There was no significant difference in age or gender distribution between smokers and non-smokers, both within each cohort and including all subjects. The additional four Belfast subjects did not differ in age (26 vs 26 years, p=0.95) or gender distribution (25% vs 47% male, p=0.41) from the initial Belfast cohort.
Alveolar-capillary membrane permeability
Alveolar-capillary membrane permeability was assessed by BAL total protein. In unstimulated BAL from the Colorado cohort, there was no significant difference in total protein between non-smokers and smokers (figure 1, see online supplementary table S1). In the Belfast cohort, after LPS inhalation, smokers had increased total protein compared with non-smokers (median: 274 vs 208 μg/mL, p=0.04) (figure 1). Linear regression demonstrated an interaction between cigarette smoking and LPS (p=0.04), indicating that cigarette smokers develop exaggerated alveolar-capillary membrane permeability compared with non-smokers in response to inhaled LPS.

Bronchoalveolar lavage (BAL) total protein in smokers versus non-smokers in an unstimulated state (Colorado) and 6 h after 50 μg lipopolysaccharide (LPS) inhalation (Belfast). Individual data points and the median value (short white bar) are shown. p, p value from a Mann–Whitney U test; p*, p value from linear regression with interaction testing between LPS and smoking for log-transformed BAL total protein.
Inflammatory biomarkers in plasma
In the Belfast cohort before LPS inhalation, there was no significant difference in plasma IL-8, IL-1β or MMP-8 in smokers compared with non-smokers. After LPS, plasma IL-8 and MMP-8 increased in both smokers and non-smokers; however, levels were higher in smokers than in non-smokers (median IL-8 level 3 vs 2 pg/mL p=0.003; median MMP-8 level 11 vs 6 ng/mL, p=0.006) (figure 2, see online supplementary table S2). There were no significant associations between smoking and plasma IL-1β or MMP-1, -2, -3, -7, or -9. Linear regression showed statistically significant interactions between cigarette smoking and LPS when examining plasma MMP-8 (p=0.045) and IL-8 (p=0.002).
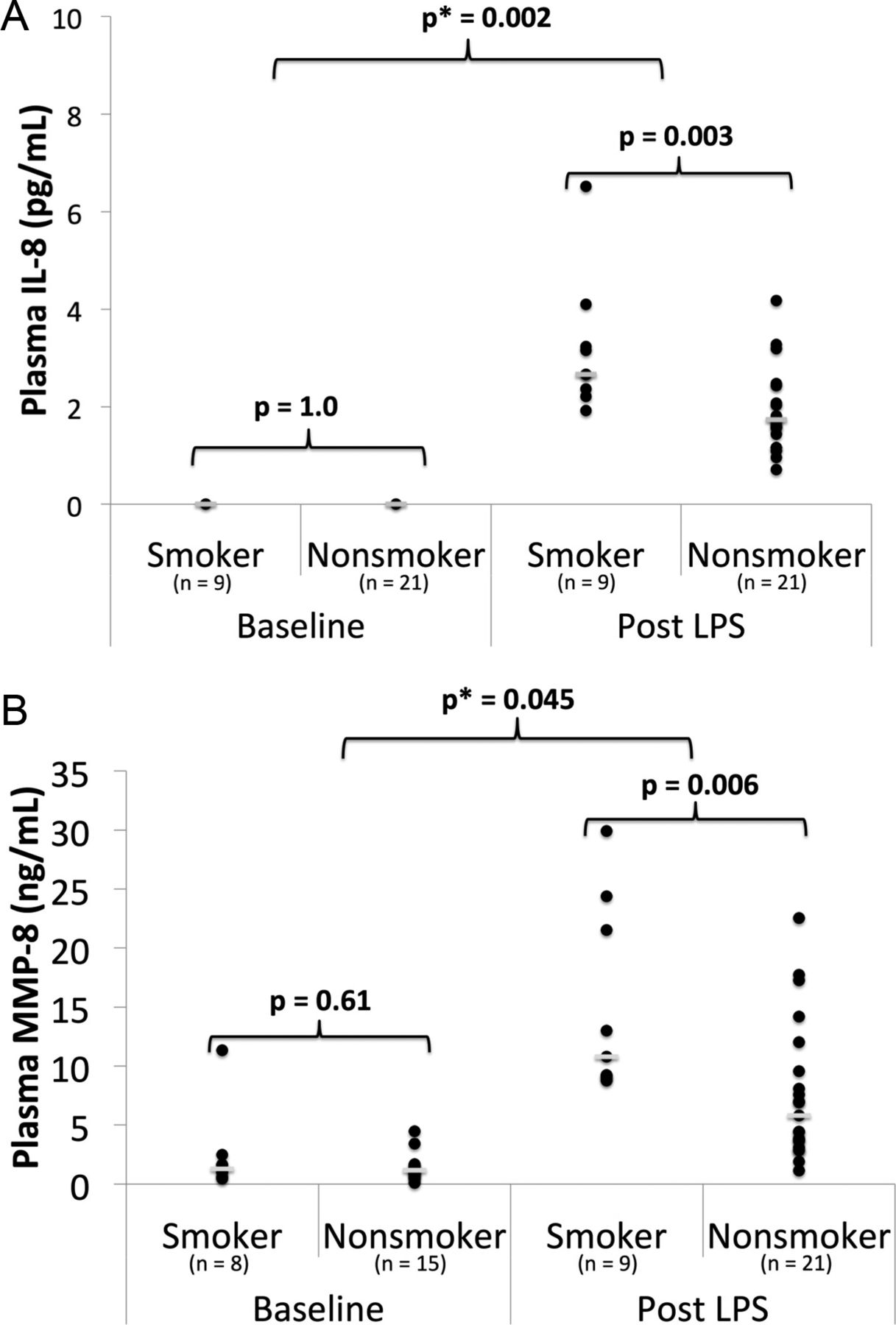
Inflammatory biomarkers in plasma in smokers versus non-smokers from the Belfast cohort at baseline and 24 h after 50 μg lipopolysaccharide (LPS) inhalation. (A) Plasma interleukin (IL)-8; (B) plasma matrix metalloproteinase (MMP)-8. Individual data points and the median value (short white bar) are shown. p, p value from a Mann–Whitney U test; p*, p value from linear regression with interaction testing between LPS and smoking for the biomarker of interest.
Inflammatory biomarkers in BAL
In unstimulated BAL from the Colorado cohort, there was no significant difference in PMNs between smokers and non-smokers, but IL-1β was higher in smokers than in non-smokers (median: 1 vs 0 pg/mL, p=0.007) (figure 3, see online supplementary table S1). In the Belfast cohort after LPS inhalation, cigarette smokers had increased BAL IL-1β (median 108 vs 37 pg/mL, p=0.002), percentage PMNs (median 42% vs 31%, p=0.02) and total PMNs (median 9 vs 3×105 cells, p=0.02) compared with non-smokers (figure 3, see online supplementary table S1). BAL IL-8 levels after LPS were similar between smokers and non-smokers. Linear regression identified significant interactions between cigarette smoking and LPS for IL-1β (p<0.001) and percentage PMNs (p=0.01), indicating that the response of these markers to LPS was significantly different in smokers compared with non-smokers.
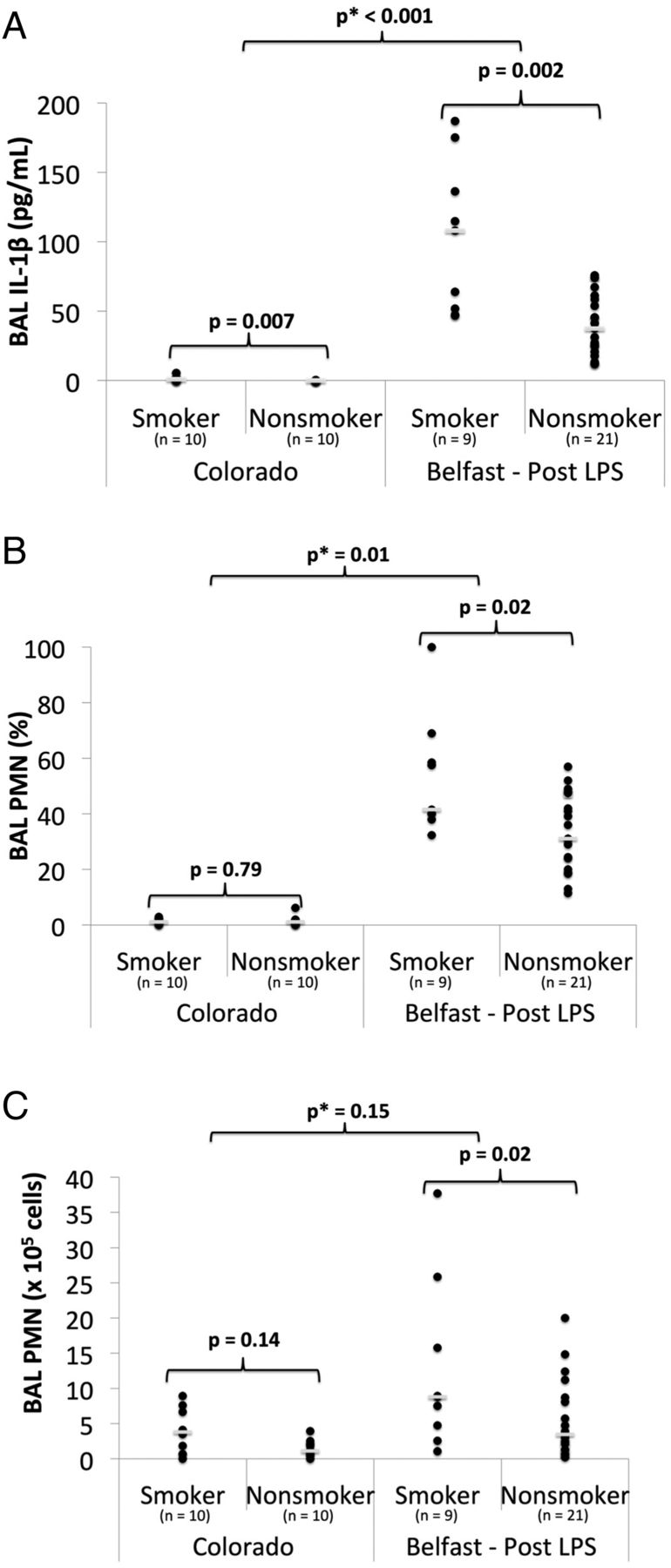
Inflammatory biomarkers in bronchoalveolar lavage (BAL) fluid of smokers versus non-smokers in an unstimulated state (Colorado) and 6 h after 50 μg lipopolysaccharide (LPS) inhalation (Belfast). (A) BAL interleukin (IL)-1β; (B) BAL polymorphonuclear neutrophil (PMN) (%); (C) BAL PMN (×105 cells). Individual data points and the median value (short white bar) are shown. p, p value from a Mann–Whitney U test; p*, p value from linear regression with interaction testing between LPS and smoking for the biomarker of interest.
SP-D in plasma
In the Belfast cohort at baseline, there was no significant difference in plasma SP-D between smokers and non-smokers. After LPS inhalation, plasma SP-D was higher (median 130 vs 96 ng/mL, p=0.04) in smokers than in non-smokers (figure 4, see online supplementary table S2). However, linear regression did not detect an interaction between LPS and smoking for plasma SP-D (p=0.31).

Surfactant protein D (SP-D) in smokers versus non-smokers. (A) Plasma SP-D from the Belfast cohort at baseline and 24 h after 50 μg lipopolysaccharide (LPS) inhalation; (B) bronchoalveolar lavage (BAL) SP-D in smokers versus non-smokers in an unstimulated state (Colorado) and 6 h after 50 μg LPS inhalation (Belfast). Individual data points and the median value (short white bar) are shown. p, p value from a Mann–Whitney U test; p*, p value from linear regression with interaction testing between LPS and smoking for plasma SP-D and log-transformed BAL SP-D.
SP-D in BAL
In unstimulated BAL from the Colorado cohort, there was no significant difference in SP-D between smokers and non-smokers (figure 4, see online supplementary table S1). In the Belfast cohort, after LPS inhalation, BAL SP-D was lower (median 724 vs 1347 ng/mL, p=0.02) in smokers than in non-smokers (figure 4). Linear regression demonstrated no interaction between cigarette smoking and LPS for BAL SP-D (p=0.13).
VEGF and receptor in plasma and BAL
There was no significant difference in plasma VEGF levels between smokers and non-smokers either at baseline or after LPS inhalation. In unstimulated BAL from the Colorado cohort, cigarette smoking was associated with decreased BAL VEGF (p=0.001) and a trend toward increased sVEGFr-1 (p=0.08) (figure 5, see online supplementary table S1). After LPS inhalation, in the Belfast cohort, cigarette smoke was associated with decreased VEGF (median 1 vs 219 pg/mL, p<0.0001) and increased sVEGFr-1 (median 99 vs 31 pg/mL, p=0.0001) in the BAL (figure 5). A test for interaction between cigarette smoke and LPS exposure was significant for both VEGF (p<0.001) and sVEGFr-1 (p<0.001), indicating that these biomarkers respond differently to LPS in smokers versus non-smokers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bronchoalveolar lavage (BAL) vascular endothelial growth factor (VEGF) and a soluble form of VEGF-1 receptor (sVEGFr-1) in smokers versus non-smokers in an unstimulated state (Colorado) and 6 h after 50 μg lipopolysaccharide (LPS) inhalation (Belfast). (A) BAL VEGF; (B) BAL sVEGFr-1. Individual data points and the median value (short white bar) are shown. p, p value from a Mann–Whitney U test; p*, p value from linear regression with interaction testing between LPS and smoking for log-transformed VEGF and sVEGFR-1.
Effect of statins and age on analyses
Since some subjects in the Belfast cohort were randomised to a statin, linear regression was used to determine whether findings were independent of statin exposure. This analysis confirmed that all findings were independent of statin use (see online supplementary tables S3 and S5). Additionally, since the Belfast and Colorado cohorts differed in age, we adjusted for age when performing interaction testing between the cohorts, and all findings were independent of age and statin use (see online supplementary tables S4 and S6).
Cohort comparison
To ensure that differences identified above were due to smoking status and not unmeasured differences in patients enrolled by the two sites, we enrolled a separate small cohort of smokers who underwent bronchoscopy both with and without LPS exposure, as described in ‘Subjects and methods’. Baseline levels of BAL biomarkers in these subjects did not significantly differ from those of the smokers from our Colorado cohort, with the exception of sVEGRr-1, which was raised in comparison with our Colorado cohort (median 21 vs 1 pg/mL, p=0.01).
Next, we compared BAL biomarkers from the four additional Belfast subjects’ first bronchoscopy (baseline) with those obtained during their second bronchoscopy (post-LPS). From baseline to post-LPS, these subjects showed dramatic increases in BAL IL-1β, IL-8 and total protein with less dramatic changes in SP-D, sVEGFr-1 and VEGF levels (see online supplementary figure S1, table S7). This pattern of changes closely mirrored those we observed in comparing smokers from our Colorado cohort with our Belfast cohort.
Discussion
This study indicates that healthy cigarette smokers have an altered response to inhaled LPS that is remarkably similar to the pattern of findings in patients with ARDS, including altered alveolar-capillary permeability to protein, as well as acute inflammation and lung epithelial cell injury. The finding that smokers are more prone than non-smokers to the development of alveolar-capillary barrier dysfunction in the presence of an inflammatory stimulus provides a key potential explanation for the epidemiological links between smoking and the development of human ARDS.
Increased alveolar-capillary membrane permeability is the fundamental pathophysiological hallmark of ARDS. The disruption of the alveolar-capillary membrane, due to endothelial and/or alveolar epithelial injury, results in the influx of protein-rich oedema fluid into the alveolar space. Patients with ARDS have raised total protein levels in oedema fluid and BAL compared with those with cardiogenic oedema.13 ,14 Furthermore, increased BAL total protein levels in patients with ARDS have been associated with poor outcomes.15 In this study, cigarette smokers developed increased alveolar-capillary membrane permeability to protein after LPS inhalation compared with non-smokers, suggesting that smokers may be more prone to developing increased barrier permeability in the presence of a ‘second hit,’ such as pneumonia or sepsis.
The remaining biomarkers in this study were selected a priori based on previous studies in patients with ARDS to determine whether the mechanisms they represent may mediate the relationship between cigarette smoke exposure and ARDS. In humans, BAL IL-1β peaks at ARDS onset16 and remains high in non-survivors.17 Similarly, IL-8 has been identified as an important mediator of lung injury in ARDS18 and is predictive of ARDS onset and associated with clinical outcomes.19 IL-1β and IL-8 are critical to the endothelial adhesion and recruitment of neutrophils into the lung airspaces, and neutrophils play a key role in the dysregulated inflammation implicated in ARDS pathogenesis.20 Neutrophils act through a variety of mechanisms, including the formation of neutrophil-extracellular traps and the release of reactive oxygen species and proteinases, including MMPs, which participate in basement membrane breakdown and are raised in patients with ARDS21 and predictive of outcomes.22
Previous studies suggest that cigarette smoke is associated with alveolar inflammation. In healthy volunteers, cigarette smoke has been associated with increased alveolar macrophages and BAL proinflammatory cytokines such as IL-1ß,6 although the association with IL-8 is more controversial.6 ,23 Furthermore, in a previous study, healthy smokers had increased BAL IL-1β and PMNs but not IL-8 compared with non-smokers after LPS inhalation,24 although no interaction testing was performed. In our study, cigarette smokers had a statistically significant exaggerated inflammatory response, as determined by interaction testing, compared with non-smokers. This finding implies that smokers respond differently to an inflammatory stimulus than non-smokers, and this exaggerated inflammatory response may contribute to the increased alveolar-capillary membrane permeability that was seen in our study. Given the importance of inflammation in ARDS pathogenesis, this exaggerated inflammatory response may play a significant role in increasing the risk of ARDS in cigarette smokers.
SP-D is a biomarker of type II epithelial cell injury. Plasma SP-D is increased in patients with ARDS25 and associated with increased mortality and fewer ventilator-free days.26 Decreased BAL SP-D is associated with increased mortality in patients with ARDS.25 Low tidal volume ventilation attenuated the rise of plasma SP-D, indicating that alveolar epithelial injury is fundamental to ARDS pathogenesis.26 In our study, smokers had more severe alveolar epithelial injury after LPS inhalation, as measured by SP-D, than non-smokers. Similar changes have also been seen in critical illness, as donor smoking was associated with decreased BAL SP-D in ex vivo human lungs rejected for organ transplantation.10 Taken together, these findings indicate that cigarette smoking is associated with early alveolar epithelial injury that may be identifiable only in the presence of a ‘second hit’. Furthermore, given the importance of the alveolar epithelium to alveolar-capillary membrane integrity, this finding may, in part, explain both the increased barrier permeability after LPS inhalation and the increased risk of ARDS in smokers.
VEGF is important for endothelial cell survival, regulating permeability and angiogenesis. In previous studies, BAL VEGF was decreased in healthy smokers,6 although plasma VEGF was unchanged.27 These effects of smoking on VEGF are thought to play a key mechanistic role in a variety of diseases, such as COPD.28 In ARDS, patients have raised plasma VEGF,29 and plasma from patients with ARDS increases permeability across endothelial cell monolayers, an effect prevented by the addition of VEGF inhibitors,29 suggesting a role for VEGF in ARDS pathogenesis. In BAL, decreased VEGF has been associated with ARDS, while a rise is associated with ARDS recovery,30 although a different study reported equivalent levels in hydrostatic and permeability oedema.31 sVEGFr-1 inhibits VEGF, and is increased in alveolar fluid from patients with ARDS.32 Our study confirms baseline VEGF homoeostatic abnormalities in the lungs of smokers, and also implies that smokers have a different response to LPS than non-smokers as regards VEGF and its natural inhibitor. As this pattern of abnormalities is similar to that of patients with ARDS, the abnormal VEGF homoeostasis in smokers may promote ARDS in the presence of a less severe stimulus, echoing clinical observations.5
This study has several strengths. By studying healthy volunteers using an experimental model of ARDS, we were able to test the effect of cigarette smoke on pathways of interest without the comorbidities and confounders typical of cohort studies. Furthermore, since chronic alcohol use is associated with an increased risk of ARDS,33 and chronic alcohol use and cigarette smoking often coexist, the exclusion of chronic alcohol users from our study eliminates a key confounder. A second strength of this study was interaction testing between cigarette smoke and LPS. Whereas previous studies have noted a variety of differences between smokers and non-smokers, our study design and use of interaction testing found that the response of cigarette smokers to LPS is statistically different from that of non-smokers. Finally, results from the four additional smokers who underwent bronchoscopy both before and after LPS supported the findings that we identified in the smokers in our two cohorts, although the small sample size of this group does limit statistical validation.
This study also has limitations. First, cigarette smoking history was obtained by self-report. Previous studies suggest that biomarkers of cigarette smoke exposure provide a more accurate assessment of true exposure in critically ill populations.34 However, since subjects in this study were healthy young volunteers, both self-reported smoking status and alcohol use should be accurate.35 ,36 Second, the modest sample size limited our power to detect differences in biomarkers between smokers and non-smokers. However, even with this sample size, the magnitude of the differences in biomarkers between smokers and non-smokers was large and statistically significant, supporting our initial hypothesis. Third, some patients in the Belfast cohort received a statin. Although we adjusted for statin use with linear regression and identified no substantive effects on our main analyses, future studies should consider eliminating this potential confounder. Finally, owing to the logistical difficulties of enrolling large numbers of subjects in a study with multiple bronchoscopy procedures, we used two cohorts to compare BAL biomarkers. Although there are potential differences between these two cohorts, we used linear regression to adjust for age and statin use, and all subjects were young and without major comorbidities. Furthermore, BAL biomarkers from the Colorado cohort were similar to those in several previous studies of healthy smokers and non-smokers,37–39 supporting our decision to use this cohort as a baseline comparison group. Lastly, BAL biomarkers before LPS exposure in the four additional Belfast smokers who underwent bronchoscopy both before and after LPS did not differ significantly from those of the Colorado cohort, with the exception of BAL sVEGFr-1. In all smokers, unstimulated BAL sVEGFr-1 levels were near the minimum detectable level of our assay. In the Colorado cohort, 5/10 smokers had undetectable levels. BAL sVEGFr-1 in the five subjects with detectable levels did not differ from unstimulated levels in the additional four Belfast subjects. These findings suggest that our decision to compare two different cohorts was reasonable but do not exclude the possibility of additional unmeasured confounders.
In conclusion, this study suggests that cigarette smoke primes the lungs to develop ARDS by promoting an abnormal response to a ‘second hit,’ with increased alveolar-membrane capillary permeability, exaggerated inflammation, alveolar epithelial injury and endothelial dysfunction. The identification of key mechanisms that may predispose smokers to the development of ARDS lays the foundation for preventative strategies that could be beneficial for smokers clinically at risk for ARDS or therapeutic strategies in smokers with ARDS. In addition, since the pattern of biomarker abnormalities in the LPS model mirrors that in ARDS and we observed smoking-associated changes in these biomarkers, LPS inhalation may serve as a new model for the identification of acute pulmonary toxicities of existing and new tobacco products, including e-cigarettes. The use of this model and the identification of biomarkers of harm associated with tobacco products may inform the risk evaluation and regulation of tobacco products. Although further research is needed to validate our findings in critically ill subjects, this study fills an important gap in our understanding of the mechanistic relationship between cigarette smoke exposure and ARDS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors FM, MAM, DFM and CSC: study conception and design; ELB, RWV, CMO, MS, UH, JA, DRT and DFM: data acquisition; FM, MAM, DFM, CSC: data analysis and interpretation. FM, MAM, DFM and CSC: drafted the manuscript. All authors participated in critically revising the manuscript, saw and approved the final version of the manuscript. FM takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding Supported by Northern Ireland R&D Office; UK Intensive Care Society (DFM); NHLBI HL51856 (MAM), HL110969 (CSC), HL126345-01 (FM) and R24 AA019661 (ELB). Some research reported in this publication was supported by grant number 1P50CA180890 from the National Cancer Institute and Food and Drug Administration Center for Tobacco Products (CSC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Food and Drug Administration.
Competing interests None declared.
Ethics approval Queen's University Belfast and University of Colorado institutional review boards.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Airwaves