Article Text

Abstract
Background Sensitisation with Aspergillus fumigatus (Af) is known to be associated with severe allergic lung inflammation, but the mechanism remains to be clarified. Phosphoinositide 3-kinase (PI3K)-δ and endoplasmic reticulum (ER) stress are suggested to be involved in steroid-resistant lung inflammation. We aimed to elucidate the role of PI3K-δ and its relationship with ER stress in fungus-induced allergic lung inflammation.
Methods Using Af-exposed in vivo and in vitro experimental systems, we examined whether PI3K-δ regulates ER stress, thereby contributing to steroid resistance in fungus-induced allergic lung inflammation. Moreover, we checked expression of an ER stress marker in lung tissues isolated from patients with allergic bronchopulmonary aspergillosis.
Results Af-exposed mice showed that ER stress markers, unfolded protein response (UPR)-related proteins, phosphorylated Akt, generation of mitochondrial reactive oxygen species (mtROS), eosinophilic allergic inflammation, and airway hyperresponsiveness (AHR) were increased in the lung. Similarly, glucose-regulated protein 78 was increased in lung tissues of patients with ABPA. A PI3K-δ inhibitor reduced Af-induced increases in ER stress markers, UPR-related proteins, allergic inflammation and AHR in mice. However, dexamethasone failed to reduce Af-induced allergic inflammation, AHR and elevation of ER stress. Administration of an ER stress inhibitor or a mtROS scavenger improved Af-induced allergic inflammation. The PI3K-δ inhibitor reduced Af-induced mtROS generation and the mtROS scavenger ameliorated ER stress. In primary cultured tracheal epithelial cells, Af-induced ER stress was inhibited by blockade of PI3K-δ.
Conclusions These findings suggest that PI3K-δ regulates Af-induced steroid-resistant eosinophilic allergic lung inflammation through ER stress.
- Allergic lung disease
- Asthma Mechanisms
- Aspergillus Lung Disease
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
Does phosphoinositide 3-kinase (PI3K)-δ play a crucial role in fungus-induced severe allergic lung inflammation?
What is the bottom line?
PI3K-δ regulates Aspergillus fumigatus-induced steroid-resistant allergic lung inflammation through modulation of endoplasmic reticulum stress.
Why read on?
Inhibition of PI3K-δ is efficacious in preclinical models of fungus-induced severe allergic lung disease and may have potential for treating patients with steroid-resistant allergic lung disorders.
Introduction
Aspergillus fumigatus (Af) is ubiquitous in normal air and the human respiratory system is constantly exposed to airborne spores of Af. Various lung disorders including invasive aspergillosis, aspergilloma, bronchial asthma and allergic bronchopulmonary aspergillosis (ABPA) are commonly associated with Af.1 Among them, ABPA and bronchial asthma are representative allergic lung inflammatory disorders related to Af. ABPA is one of the complex hypersensitivity reactions to Aspergillus species usually colonised in bronchi and it occurs in patients with asthma.2 Although many patients with ABPA respond well to treatment with systemic steroids, the condition of some patients is poorly controlled by conventional management.3 Additionally, in patients with bronchial asthma, sensitisation with Af is known to cause severe phenotypes of the disease.4 ,5 However, the mechanisms by which Af is involved in allergic lung inflammation remain poorly understood.
Class I phosphoinositide 3-kinases (PI3Ks) are involved in the induction of T helper type 2 cell (TH2)-related immune responses.6 They exist as heterodimeric complexes in which a catalytic p110 subunit (designated as α, β, γ or δ) is associated with a particular regulatory subunit (designated as p85, p55, p50 or p101). These proteins are a family of lipid signalling kinases that generate phosphatidylinositol-3,4,5-trisphosphate (PIP3) and phosphorylate effector proteins such as Akt.7 The p110-α and p110-β isoforms are ubiquitously expressed. However, expression of p110-δ isoform is restricted to circulating haematogenous cells and endothelial cells.8 PI3K-δ plays a role in a variety of immune processes involving B-cell and T-cell activation,9 mast cell degranulation,10 and trafficking and activation of neutrophils and eosinophils.11 ,12 In particular, PI3K-δ mediates steroid resistance in chronic respiratory disorders in which neutrophilic inflammation predominates.13 Therefore, inhibition of this pathway may serve as a critical target for treating steroid-resistant severe asthma and non-severe asthma.14 ,15 Unfortunately, there is a lack of information on whether PI3K-δ contributes to fungus-induced steroid-resistant eosinophilic allergic lung inflammation.
The endoplasmic reticulum (ER) serves as a protein-folding factory essential for production of functional proteins and trafficking of secretory and biological membrane proteins in cells.16 ER stress causes accumulation of unfolded and/or misfolded proteins in ER, interferes with protein synthesis and secretion, induces reactive oxygen species (ROS) generation, and increases inflammation partly via nuclear factor (NF)-κB activation.17 As a result, ER stress is implicated in the pathogenesis of diverse inflammatory processes.18 Recently, our group demonstrated that ER stress influences the pathogenesis of bronchial asthma, especially the steroid-resistant neutrophilic asthma model.19 Although various signalling pathways are involved in ER stress,20 ,21 the relationship between PI3K-δ and ER stress in allergic lung inflammation is not clearly understood. In addition, there are few data about the role of ER stress in fungus-induced allergic lung inflammation.
In this study, we examined whether PI3K-δ regulates ER stress, thereby contributing to steroid resistance in eosinophil-dominant allergic lung inflammation using Af-exposed mice and primary cultured tracheal epithelial cells (TECs). In addition, we analysed the expression of a representative ER stress marker, glucose-regulated protein 78 (GRP78), in lung tissues from patients with ABPA to evaluate the association of ER stress with the pathogenesis of Af-related lung inflammation in humans.
Methods
Animals and experimental protocol
Female C57BL/6 mice, 8–10 weeks of age and free of murine specific pathogens, were used. All experimental animals used in this study were under a protocol approved by the Institutional Animal Care and Use Committee of the Chonbuk National University (CBU 2014-00030). For the generation of an Af-induced allergic lung inflammation model, mice were sensitised and challenged with Af antigen, in which the cellular fungal material was inactivated and lyophilised, as previously described.22 Bronchoalveolar lavage (BAL) was performed at 72 h after the last challenge with Af in Af sensitised and challenged mice.
Administration of drugs
A selective p110-δ inhibitor, IC87114 (0.1 or 1 mg/kg body weight/day, Calbiochem, San Diego, California, USA) or vehicle control (0.05% dimethyl sulfoxide, DMSO) diluted with 0.9% NaCl was administered once by intratracheal instillation to each animal 24 h after the last challenge with Af. A chemical chaperone, 4-phenylbutyric acid (4-PBA) (80 mg/kg body weight/day, Calbiochem) diluted with phosphate-buffered saline, was administered twice by intraperitoneal injection to each animal 24 h before and after the last challenge with Af. An inhibitor of NF-κB activation, BAY 11-7085 (20 mg/kg body weight/day, BIOMOL International L.P., Plymouth Meeting, Pennsylvania, USA) dissolved in DMSO and diluted with 0.9% NaCl was administered twice by intraperitoneal injection to each animal 24 h before and after the last challenge with Af. Dexamethasone (1 mg/kg body weight/day, Sigma-Aldrich, St Louis, Missouri, USA) diluted with saline was administered twice by means of oral gavage to each animal, once 24 h before and again at 2 h before the last challenge with Af.19 ,23 A mitochondrial ROS (mtROS) scavenger, NecroX-5 (30 mg/kg body weight/day, Enzo Life Sciences, Farmingdale, New York, USA) diluted with distilled water was administered twice by intraperitoneal injection to each animal 24 h before and after the last challenge with Af.
Measurement of PI3K enzyme activity in lung tissue
The amount of PIP3 produced was quantified by the PIP3 competition enzyme immunoassay (Echelon, Inc., Salt Lake City, Utah, USA).
Measurement of glutathione and glutathione disulfide in lung tissues
Total glutathione (GSH) and glutathione disulfide (GSSG) levels were determined using a GSH Assay Kit (Cayman Chemical Co., Ann Arbor, Michigan, USA) according to the manufacturer's protocol.
Serum total IgE and Af-specific IgE
For the Af-specific IgE assay, 96-well immunosorbent plates were coated with Af antigen (10 μg/mL, Greer Laboratories, Lenoir, North Carolina, USA). Horseradish peroxidase-conjugated goat anti-mouse IgE antibody (Bethyl Laboratories, Montgomery, Texas, USA) was used to detect Af-bound IgE. Total serum IgE was measured using a mouse Total IgE ELISA Kit (MD Bioproducts, St Paul, Minnesota, USA) according to the manufacturer's protocol.
Isolation and primary culture of murine TECs and IC87114 treatment
Murine TECs were isolated under sterile conditions as described previously.6 To verify the role of PI3K-δ in the regulation of ER stress in Af-stimulated primary cultured TECs, cells were treated with IC87114 (10 μmol/L) for 2 h, and then stimulated by Af antigen (5 μg/mL) for an additional 12 h.
PI3K-δ specific or scrambled small interfering RNA transfection in primary cultured TECs
Primary cultured murine TECs were transfected with the PI3K-δ specific (PIK3CD) or scrambled small interfering RNA (siRNA) (Dharmacon, Lafayette, Louisiana, USA) by using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, California, USA).
Immunofluorescence staining for GRP78, CCAAT/enhancer-binding protein-homologous protein (CHOP) and mtROS
Immunofluorescence staining for GRP78 and CHOP was performed as previously described.19 To demonstrate the intensity of mtROS in Af-stimulated primary cultured TECs and BAL cells, cells were stained with Mitotracker Red CM-H2ROS (Invitrogen) and analysed using a confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
Western blot analysis
Protein levels were determined by Western blotting as previously described.19
Nuclear protein extractions
Extraction of nuclear proteins was performed as previously described.19
Histology
Histologic assessment was performed as previously described.19
Determination of airway responsiveness to methacholine
Invasive measurement of airway responsiveness was performed as described elsewhere.19
Statistics
We used SPSS statistical software (V.18.0, SPSS, Chicago, Illinois, USA). Data are expressed as mean±SEM. Statistical comparisons were performed using one-way ANOVA followed by Scheffe's test. The Mann–Whitney U test was used to compare the two groups of human subjects. A value of p<0.05 was considered statistically significant.
Results
PI3K-δ regulates ER stress in vivo
To examine whether p110-δ contributes to regulation of ER stress in Af-exposed mice, we evaluated the effects of IC87114, a selective p110-δ inhibitor, on levels of ER stress markers, GRP78 and CHOP, and unfolded-protein response (UPR)-related markers in lung tissues. Levels of GRP78 and CHOP were substantially increased in Af-exposed mice compared with those in control mice (figure 1A–D). Af-induced increases of GRP78 and CHOP were significantly reduced by administration of IC87114.

Inhibition of phosphoinositide 3-kinase (PI3K)-δ reduces endoplasmic reticulum (ER) stress and unfolded protein response (UPR) in Aspergillus fumigatus (Af)-exposed mice. Representative immunoblots of GRP78 (A), CHOP (C), phospho (p)-IRE1α (E), p-eIF2α (G), XBP-1 (I) and ATF-4 (K) in lung tissues and densitometric analyses of GRP78 (B), CHOP (D), p-IRE1α (F), p-eIF2α (H), XBP-1 (J) and ATF-4 (L) from saline sensitised and challenged mice administered drug vehicle (dimethyl sulfoxide, DMSO) (SAL+DMSO), Af sensitised and challenged mice administered drug vehicle (Af+DMSO), Af sensitised and challenged mice administered 0.1 mg/kg IC87114 (Af+IC 0.1) or Af sensitised and challenged mice administered 1.0 mg/kg IC87114 (Af+IC 1.0). Bars represent mean±SEM from six mice per group (five mice per group for p-eIF2α). #p<0.05 versus SAL+DMSO; *p<0.05 versus Af+DMSO.
Moreover, Af-induced increases in UPR-related markers including p-IRE1α, p-eIF2α, XBP-1 and ATF-4 in Af-exposed mice were significantly reduced by administration of IC87114 (figure 1E–L).
To demonstrate the involvement of ER stress in Af-related allergic lung disorder in humans, we performed immunofluorescence and immunohistochemical analyses with lung tissues obtained from patients with ABPA and healthy controls. The results showed that GRP78 was remarkably increased in patients with ABPA compared with that in healthy controls (see online supplementary figure S1A–C).
PI3K-δ regulates ER stress in vitro
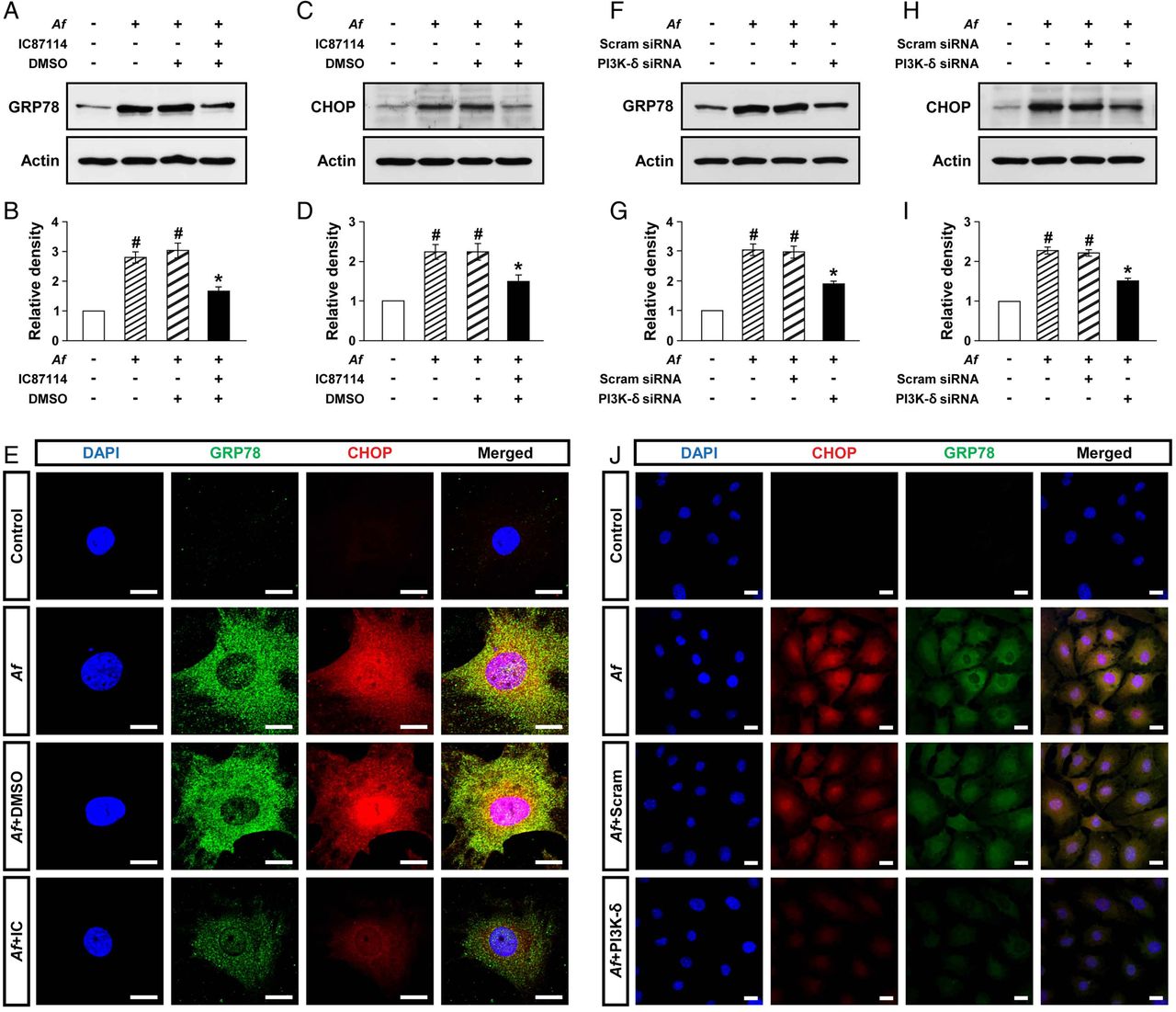
Epithelial cells are central in allergic lung inflammation through regulation of TH2-related mediators.24 In particular, we have previously shown that PI3K-δ is involved in the pathogenesis of allergic lung disorders through modulating the release of mediators in the lung epithelial cells.25 Therefore, to further assess the above interrelationship between PI3K-δ and ER stress in Af-induced allergic lung inflammation, primary cultured TECs were used. Levels of GRP78 and CHOP were significantly increased at 12 h after Af stimulation compared with the levels in control cells (figure 2A–D). Treatment with IC87114 significantly reduced Af-induced increases in GRP78 and CHOP. Supporting the data, treatment with IC87114 limited Af-induced increases in immunofluorescence intensities of GRP78 and CHOP in these cells (figure 2E).

Phosphoinositide 3-kinase (PI3K)-δ modulates endoplasmic reticulum (ER) stress in vitro. Representative immunoblots of GRP78 (A) and CHOP (C) after stimulation with Aspergillus fumigatus (Af) in primary cultured murine tracheal epithelial cells (TECs) in the presence and absence of IC87114 and densitometric analyses of GRP78 (B) or CHOP (D). Bars represent mean±SEM from five independent experiments. #p<0.05 versus control; *p<0.05 versus cells stimulated with Af alone. (E) Representative confocal laser immunofluorescence photomicrographs of primary cultured TECs showed the localisation of GRP78 and CHOP in controls (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered drug vehicle (dimethyl sulfoxide, DMSO) (Af+DMSO), or Af-stimulated cells administered IC87114 (Af+IC). 4′,6-diamidino-2-phenylindole (DAPI) stain was used for nuclear localisation. The right panels show the merger views. Bars indicate 20 μm. Representative immunoblots of GRP78 (F) and CHOP (H) after stimulation with Af in primary cultured murine TECs transfected with either scrambled small interfering RNA (Scram siRNA) or PI3K-δ-specific siRNA and densitometric analyses of GRP78 (G) or CHOP (I). Bars represent mean±SEM from six independent experiments. #p<0.05 versus control; *p<0.05 versus cells stimulated with Af transfected with scrambled siRNA. (J) Representative confocal laser immunofluorescence photomicrographs of primary cultured TECs showed the localisation of CHOP and GRP78 in controls (no treatment), Af-stimulated cells (Af), Af-stimulated cells administered scrambled siRNA (Af+Scram), or Af-stimulated cells administered PI3K-δ-specific siRNA (Af+PI3K-δ). Bars indicate 20 μm.
To support the link of PI3K-δ to ER stress, PI3K-δ was knocked down in TECs using PI3K-δ-specific siRNA. The level of PI3K-δ mRNA was remarkably increased at 12 h after Af stimulation compared with control cells, suggesting that Af regulates PI3K-δ expression at gene level (see online supplementary figure S2). In addition, Af-induced increase in PI3K-δ mRNA was significantly decreased by transfection of PI3K-δ-specific siRNA. Moreover, Af-induced increases in levels of GRP78 and CHOP were significantly decreased by PI3K-δ-specific siRNA (figure 2F–I). Similarly, Af-induced increases in immunofluorescence intensities of GRP78 and CHOP were decreased by PI3K-δ-specific siRNA (figure 2J). However, no significant changes were observed by scrambled siRNA.
IC87114 inhibits PI3K activity
To ensure an involvement of PI3K-δ in Af-induced allergic lung inflammation, we measured levels of p-Akt and PI3K activity in the lung. The level of p-Akt protein was increased at 72 h after Af challenge compared with the level in control mice (figure 3A, B). Consistent with these findings, PI3K activity was significantly increased after Af challenge compared with that in control mice (figure 3C). Intratracheal administration of IC87114 reduced Af-induced increases of p-Akt and PI3K activity. However, no significant changes in total Akt levels were observed in any of the groups tested.

Inhibition of phosphoinositide 3-kinase (PI3K)-δ ameliorates Aspergillus fumigatus (Af)-induced allergic lung inflammation. Representative immunoblots of p-Akt and total (t)-Akt in lung tissues (A) from SAL+dimethyl sulfoxide (DMSO), Af+DMSO, Af+IC 0.1 or Af+IC 1.0, and densitometric analyses of p-Akt (B). Bars represent mean±SEM from six mice per group. (C) Enzyme immunoassays of PIP3 in lung tissues. Bars represent mean±SEM from six mice per group. (D) Cellular changes in bronchoalveolar lavage (BAL) fluids. Bars represent mean±SEM from six mice per group. (E) Airway responsiveness assessed by invasive (Rrs) measurements. Bars represent mean±SEM from five mice per group. Representative H&E stained sections of the lung from SAL+DMSO (F), Af+DMSO (G), Af+IC 0.1 (H) or Af+IC 1.0 (I). Bars indicate scale of 50 μm. Representative immunoblots of IL-4 (J), IL-5 (L), IL-13 (N) and IL-17 (P) in lung tissues and densitometric analyses of IL-4 (K), IL-5 (M), IL-13 (O) and IL-17 (Q). Bars represent mean±SEM from six mice per group. (R) Levels of total IgE and Af-specific IgE in the serum. Bars represent mean±SEM from five mice per group. #p<0.05 versus SAL+DMSO; *p<0.05 versus Af+DMSO.
Effect of IC87114 on Af-induced allergic lung inflammation
An increase in the numbers of cells, especially eosinophils, in BAL fluids of Af-exposed mice was significantly reduced by administration of IC87114 (figure 3D).
Airway hyperresponsiveness (AHR) was assessed as a change in respiratory system resistance (Rrs). In Af-exposed mice, the dose–response curve of Rrs shifted to the left compared with that in control mice (figure 3E). Administration of IC87114 at 1 mg/kg substantially reduced Rrs at 25 and 50 mg/mL of methacholine.
Histological analysis revealed infiltration of various inflammatory cells into bronchioles of Af-exposed mice (figure 3G) compared with those in control mice (figure 3F). In addition, Af-exposed mice treated with 0.1 mg/kg (figure 3H) or 1.0 mg/kg (figure 3I) of IC87114 showed marked reduction in infiltration of inflammatory cells.
We determined levels of TH2 cytokines including IL-4, IL-5, and IL-13 and IL-17 in the lung. Western blot analyses showed that Af-induced increase in these cytokines was significantly reduced by administration of IC87114 (figure 3J–Q).
Lastly, we determined whether IC87114 modifies Af-specific TH2 response by analysing IgE levels. Treatment with IC87114 significantly lowered Af-induced increases in circulating total and Af-specific IgE level in a dose-dependent manner (figure 3R).
Effects of dexamethasone on pathophysiologic features and ER stress in Af-induced allergic lung inflammation
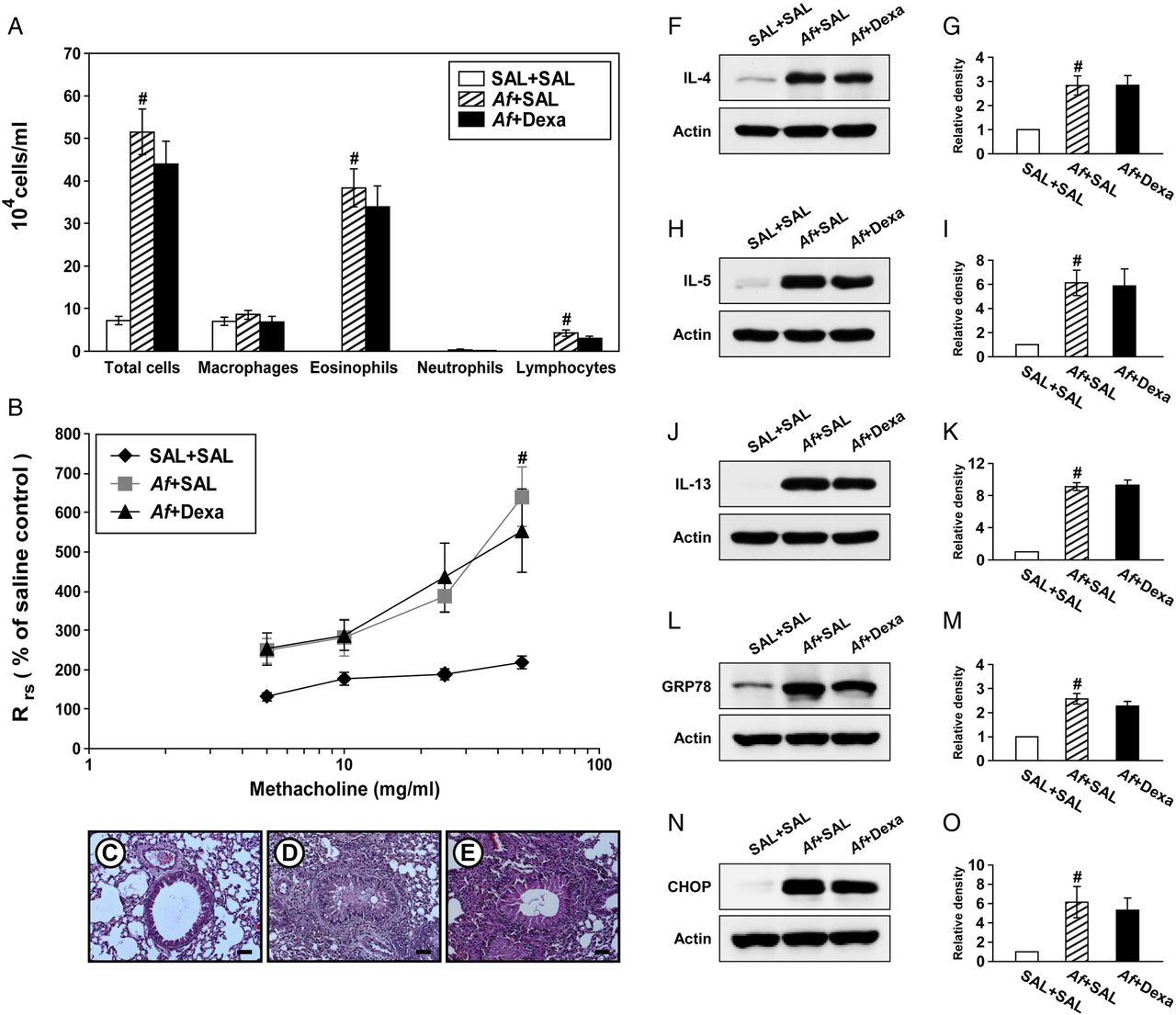
Steroid resistance is a common feature in severe allergic inflammation.15 Therefore, we investigated whether dexamethasone improves pathophysiologic features of Af-induced allergic lung inflammation. Increases in the numbers of cells including eosinophils in BAL fluids from Af-exposed mice were not significantly changed by treatment with dexamethasone (figure 4A). In addition, treatment with dexamethasone did not improve the increase in Rrs induced by methacholine (figure 4B). In Af-exposed mice administered dexamethasone, the grade of inflammatory cell infiltration into bronchioles was similar to that of Af-exposed mice administered the drug vehicle only (figure 4C–E). Moreover, increases in levels of IL-4, IL-5 and IL-13 in the lung were not significantly reduced by dexamethasone (figure 4F–K). These results suggest that the current fungus-induced allergic inflammation model represents a steroid-resistant severe phenotype.
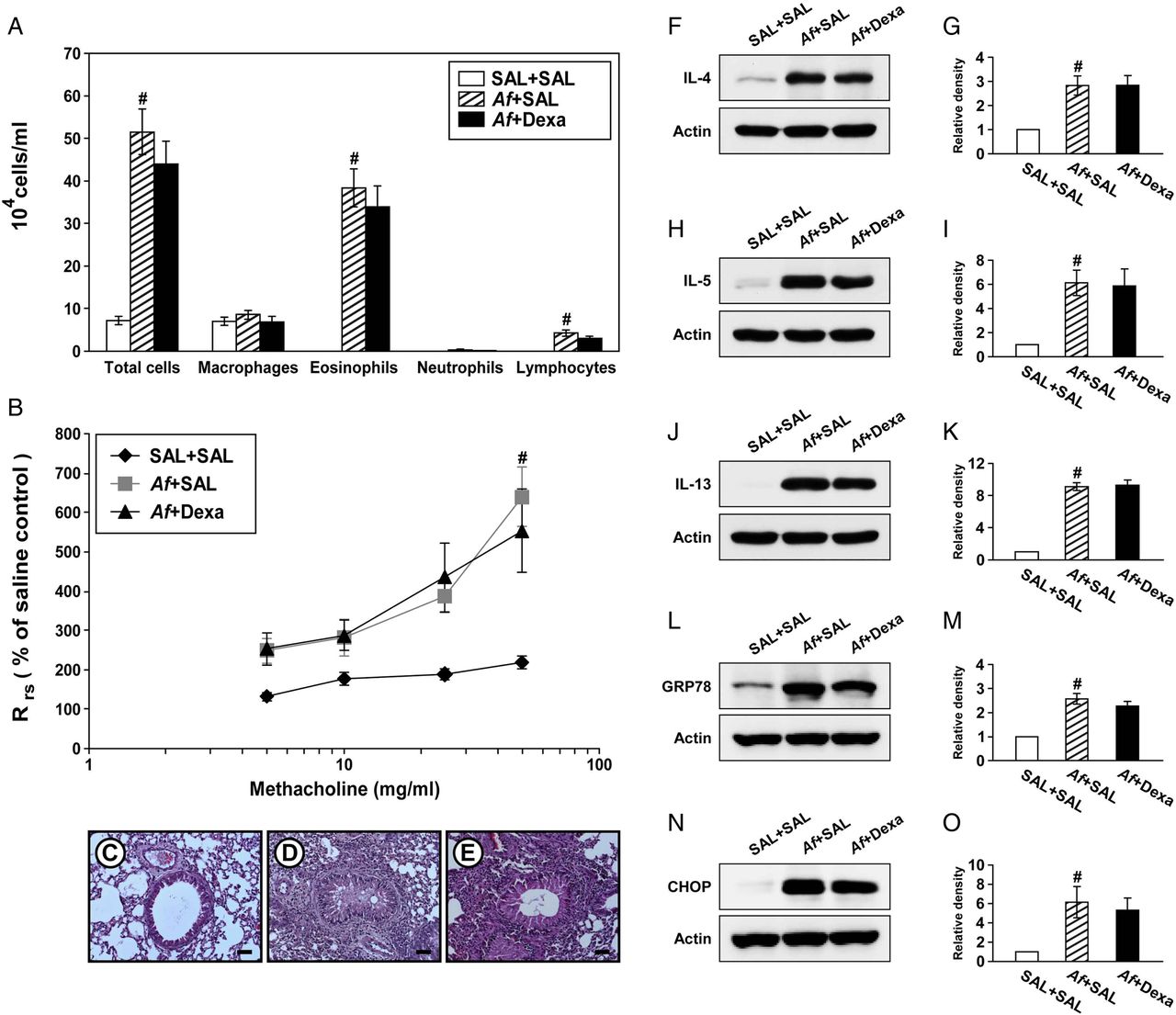
Dexamethasone treatment does not improve Aspergillus fumigatus (Af)-induced allergic inflammation and endoplasmic reticulum (ER) stress. (A) Cellular changes in bronchoalveolar lavage (BAL) fluids from saline sensitised and challenged mice administered drug vehicle (saline) (SAL+SAL), Af sensitised and challenged mice administered drug vehicle (Af+SAL) or Af sensitised and challenged mice administered dexamethasone (Af+Dexa). Bars represent mean±SEM from 10 mice per group. (B) Airway responsiveness. Bars represent mean±SEM from six mice per group. Representative H&E stained sections of the lung from SAL+SAL (C), Af+SAL (D) or Af+Dexa (E). Bars indicate scale of 50 μm. Representative immunoblots of IL-4 (F), IL-5 (H), IL-13 (J), GRP78 (L) and CHOP (N) in lung tissues and densitometric analyses of IL-4 (G), IL-5 (I), IL-13 (K), GRP78 (M) and CHOP (O) from SAL+SAL, Af+SAL and Af+Dexa. Bars represent mean±SEM from nine mice per group (five and six mice per group for GRP78 and CHOP, respectively). #p<0.05 versus SAL+SAL.
We then examined whether treatment with dexamethasone reduces Af-induced increases in ER stress markers. Interestingly, treatment with dexamethasone failed to reduce Af-induced elevation of GRP78 and CHOP in the lung of Af-exposed mice (figure 4L–O).
Inhibition of ER stress alleviates Af-induced allergic lung inflammation
To ascertain the association of ER stress with pathophysiologic features of Af-induced allergic lung inflammation, we evaluated the therapeutic effects of a potent ER stress inhibitor, 4-PBA. Increased numbers of cells, especially eosinophils, in BAL fluids of Af-exposed mice were reduced by administration of 4-PBA (figure 5A). Treatment with 4-PBA also reduced the Af-induced increase in Rrs by methacholine (figure 5B). In addition, Af-exposed mice treated with 4-PBA manifested marked reduction of the Af-induced infiltration of inflammatory cells into peribronchiolar regions (figure 5C–E).

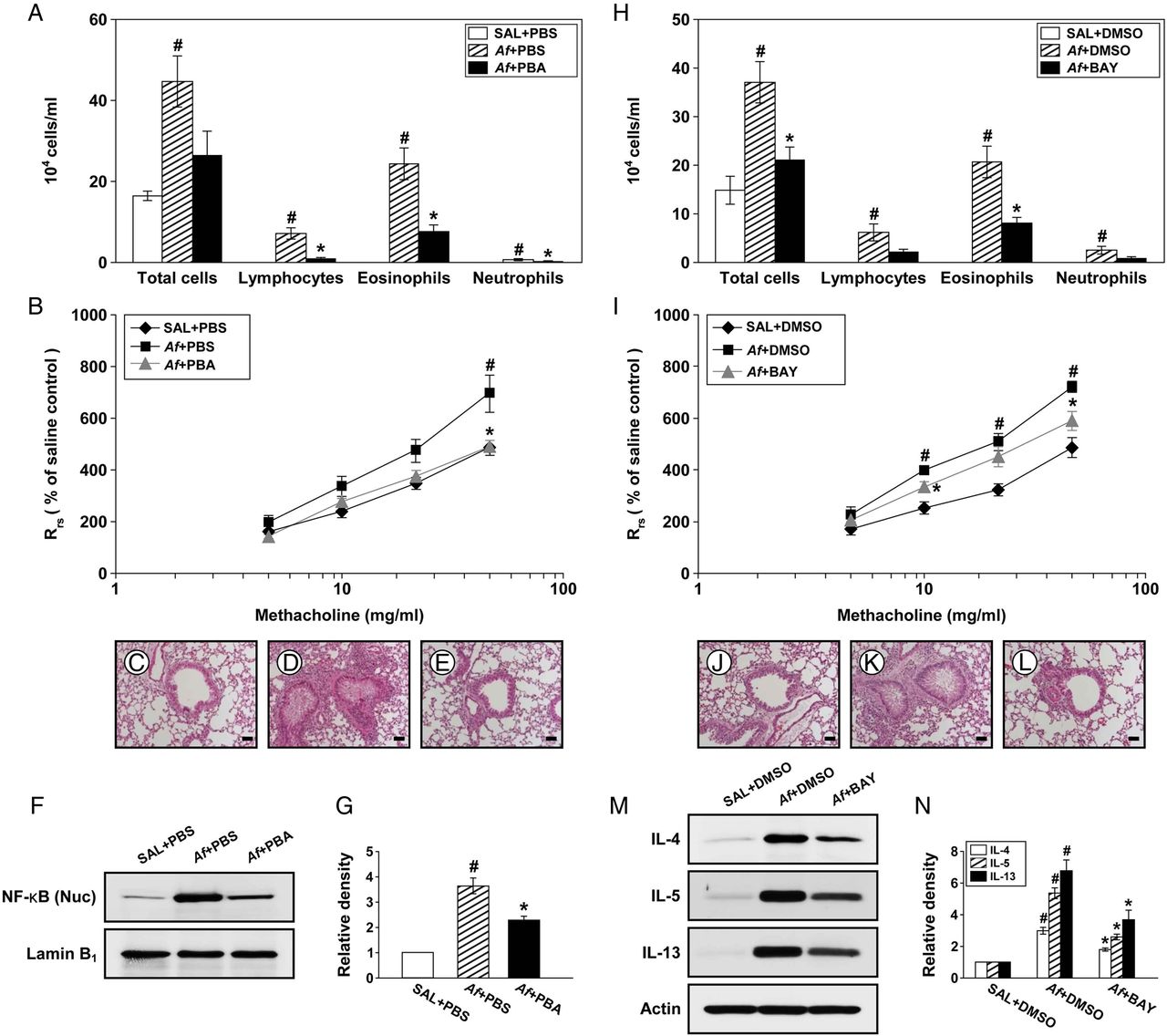
Endoplasmic reticulum (ER) stress is implicated in Aspergillus fumigatus (Af)-induced allergic lung inflammation through activating nuclear factor (NF)-κB. (A) Cellular changes in bronchoalveolar lavage (BAL) fluids from saline sensitised and challenged mice administered drug vehicle (phosphate-buffered saline, PBS) (SAL+PBS), Af sensitised and challenged mice administered drug vehicle (Af+PBS) or Af sensitised and challenged mice administered 4-phenylbutyric acid (PBA) (Af+PBA). Bars represent mean±SEM from six mice per group. (B) Airway responsiveness. Bars represent mean±SEM from five mice per group. Representative H&E stained sections of the lung from SAL+PBS (C), Af+PBS (D) or Af+PBA (E). Bars indicate scale of 50 μm. Representative immunoblot of NF-κB p65 in nuclear extracts from lung tissues (F) and densitometric analysis of NF-κB p65 (G). Bars represent mean±SEM from six mice per group. #p<0.05 versus SAL+PBS; *p<0.05 versus Af+PBS. (H) Cellular changes in BAL fluids from saline sensitised and challenged mice administered drug vehicle (dimethyl sulfoxide, DMSO) (SAL+DMSO), Af sensitised and challenged mice administered drug vehicle (Af+DMSO) or Af sensitised and challenged mice administered with BAY 11-7085 (Af+BAY). Bars represent mean±SEM from six mice per group. (I) Airway responsiveness. Bars represent mean±SEM from five mice per group. Representative H&E stained sections of the lung from SAL+DMSO (J), Af+DMSO (K) or Af+BAY (L). Bars indicate scale of 50 μm. Representative immunoblots of IL-4, IL-5 and IL-13 (M) in lung tissues and densitometric analyses of IL-4, IL-5 and IL-13 (N). Bars represent mean±SEM from six mice per group. #p<0.05 versus SAL+DMSO; *p<0.05 versus Af+DMSO.
4-PBA reduces nuclear translocation of NF-κB p65
To understand the molecular basis upon which inhibition of ER stress improves Af-induced allergic lung inflammation, we measured NF-κB p65 protein, a well known molecule involved in inflammation, in Af-exposed mice. The level of NF-κB p65 in nuclear protein extracts was significantly increased at 72 h after the last Af challenge compared with the control (figure 5F, G). The Af-induced increase in NF-κB p65 was decreased by administration of 4-PBA.
Effect of NF-κB inhibition on pathophysiologic features of Af-induced allergic lung inflammation
To further demonstrate the involvement of NF-κB, we evaluated the effects of a NF-κB inhibitor, BAY 11-7085, on Af-induced allergic lung inflammation.
Af-induced increases in numbers of total cells and eosinophils in BAL fluids were significantly reduced by administration of BAY 11-7085 (figure 5H). Treatment with BAY 11-7085 also attenuated the Rrs produced at 10 and 50 mg/mL of methacholine in Af-exposed mice (figure 5I). In addition, increases in numbers of infiltrated cells around bronchioles after the last Af challenge were markedly reduced by BAY 11-7085 (figure 5J–L). Furthermore, Af-induced increases in IL-4, IL-5 and IL-13 protein were substantially reduced by BAY 11-7085 (figure 5M, N).
IC87114 restores GSH
Oxidative stress is one of the most important features in allergic lung inflammation.26 Therefore, we measured GSH and GSSG in lung tissues. Af-exposed mice showed significantly lower GSH level than control mice (figure 6A). In contrast, GSSG level was increased after Af challenge (figure 6B). Af-induced decrease in GSH and increase in GSSG were substantially restored by administration of IC87114.

Phosphoinositide 3-kinase (PI3K)-δ regulates Aspergillus fumigatus (Af)-induced generation of mitochondrial reactive oxygen species (mtROS). Levels of glutathione (GSH) (A) and glutathione disulfide (GSSG) (B) in lung tissues from SAL+dimethyl sulfoxide (DMSO), Af+DMSO, Af+IC 0.1 or Af+IC 1.0. Bars represent mean±SEM from seven mice per group. #p<0.05 versus SAL+DMSO; *p<0.05 versus Af+DMSO. (C) Representative confocal laser immunofluorescence photomicrographs showed the localisation of mtROS in bronchoalveolar lavage (BAL) cells. Bars indicate a scale of 20 μm. (D) Representative confocal laser immunofluorescence photomicrographs of primary cultured TECs showed the localisation of mtROS in the control (no treatment), Af, Af+DMSO or Af+IC. Bars indicate a scale of 20 μm.
IC87114 reduces Af-induced mtROS generation
Mitochondrion has been recognised as a major source of intracellular ROS.27 We therefore examined the relationship between PI3K-δ and mtROS generation in our experimental systems. Immunofluorescence intensity of mtROS in BAL cells was markedly increased in Af-exposed mice compared with the intensity in control mice (figure 6C). Administration of IC87114 reduced Af-induced increase in immunofluorescence intensity of mtROS. Consistent with in vivo data, Af-induced increase in fluorescence intensity of mtROS in TECs was markedly inhibited by administration of IC87114 (figure 6D). These results indicate that PI3K-δ may induce mtROS generation.
NecroX-5 ameliorates inflammatory cell infiltration and AHR
To further investigate the role of mtROS in Af-induced allergic lung inflammation, we evaluated the treatment effects of a potent mtROS scavenger, NecroX-5.28 Increased numbers of cells, particularly eosinophils, in BAL fluids from Af-exposed mice were remarkably reduced by administration of NecroX-5 (figure 7A). In addition, Af-exposed mice treated with NecroX-5 showed marked reduction in infiltration of inflammatory cells into the peribronchiolar region compared with that in control mice (figure 7B–D). Moreover, administration of NecroX-5 resulted in a significant attenuation in Rrs at 50 mg/mL of methacholine (figure 7E).
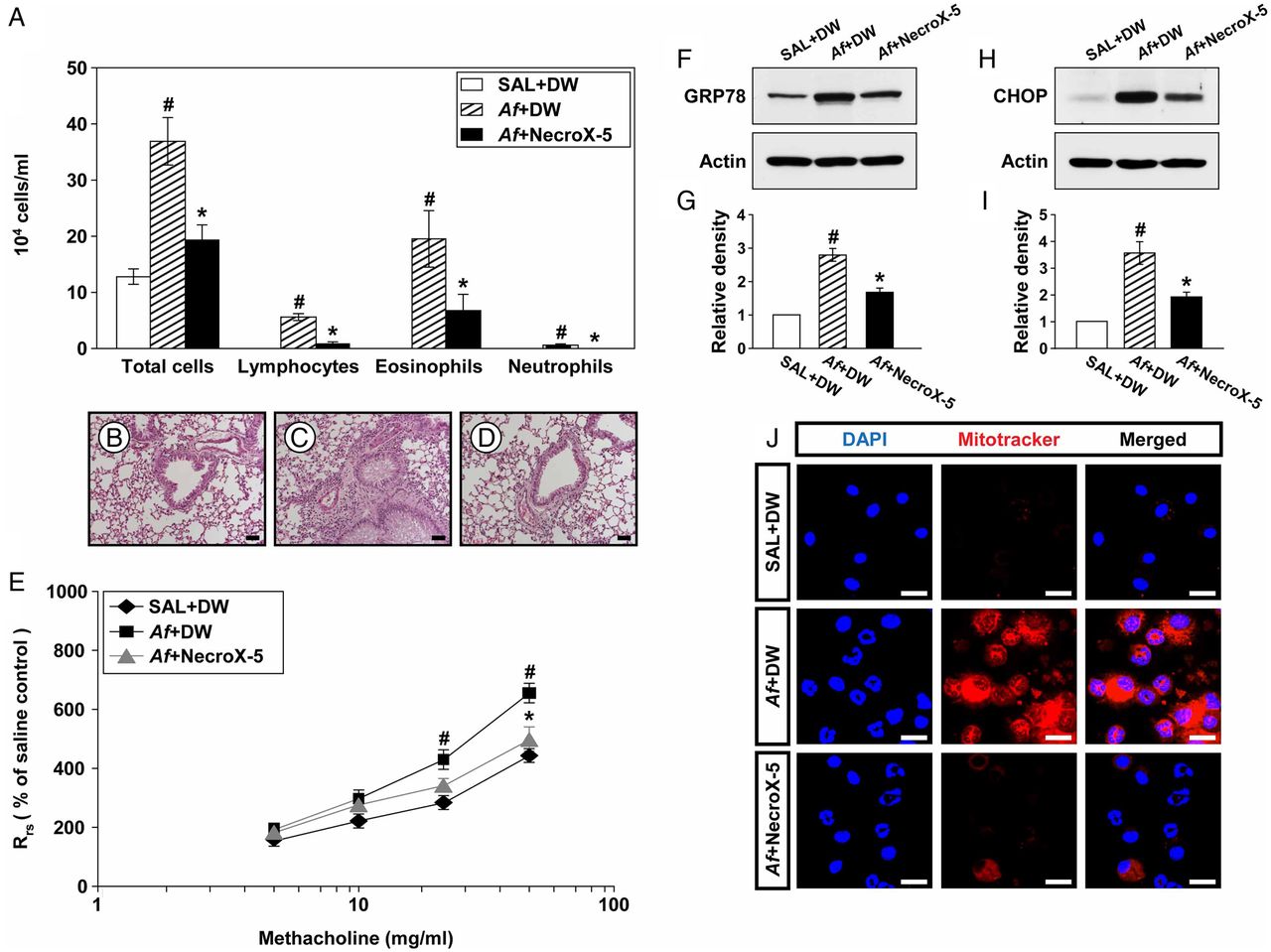
Depletion of mitochondrial reactive oxygen species (mtROS) improves Aspergillus fumigatus (Af)-induced allergic inflammation through modulating endoplasmic reticulum (ER) stress. (A) Cellular changes in bronchoalveolar lavage (BAL) fluids from saline sensitised and challenged mice administered drug vehicle (distilled water, DW) (SAL+DW), Af sensitised andchallenged mice administered drug vehicle (Af+DW), or Af sensitised and challenged mice administered NecroX-5 (Af+NecroX-5). Bars represent mean±SEM from six mice per group. Representative H&E stained sections of the lung from SAL+DW (B), Af+DW (C) and Af+NecroX-5 (D). Bars indicate scale of 50 μm. (E) Airway responsiveness. Bars represent mean±SEM from five mice per group. Representative immunoblots of GRP78 (F) and CHOP (H) in lung tissues and densitometric analyses of GRP78 (G) and CHOP (I). Bars represent mean±SEM from six mice per group. #p<0.05 versus SAL+DW; *p<0.05 versus Af+DW. (J) Representative confocal laser immunofluorescence photomicrographs showed the localisation of mtROS in BAL cells from SAL+DW, Af+DW or Af+NecroX-5. Bars indicate a scale of 20 μm.
NecroX-5 reduces GRP78 and CHOP
To investigate whether mtROS modulates ER stress, we evaluated the interrelationship between them using NecroX-5. Levels of GRP78 and CHOP were significantly elevated in Af-exposed mice compared with those in control mice (figure 7F–I). Interestingly, administration of NecroX-5 markedly lowered Af-induced increases in these proteins, suggesting that mtROS generation induces ER stress.
Effect of NecroX-5 on Af-induced mtROS generation in BAL fluids
Immunofluorescence intensity of mtROS in BAL cells was markedly increased in Af-exposed mice compared with that in control mice (figure 7J). Administration of NecroX-5 substantially reduced Af-induced increase in immunofluorescence intensity of mtROS.
Discussion
In this study, ER stress was increased in the lung of Af-exposed mice. Expression of GRP78 was also elevated in lung tissue from patients with ABPA, a hypersensitivity disorder induced by Af, compared with that in healthy controls. These results suggest that ER stress may be involved in the pathogenesis of Af-related allergic lung disorders. Additionally, we found a novel mechanism of action of PI3K-δ as an upstream regulator of ER stress through mtROS generation in Af-induced allergic lung inflammation. Also, blockade of PI3K-δ remarkably ameliorated Af-induced steroid-resistant allergic lung inflammation. These findings suggest that PI3K-δ plays an essential role in the pathogenesis of fungus-induced severe allergic lung inflammation.
Sensitivity to fungi has been known as one of the crucial determinants complicating clinical course of bronchial asthma.4 Among various species, Af is a major indoor and outdoor fungi.1 Allergic sensitisation to Af in patients with bronchial asthma is often associated with reduced lung function and immunologically mediated structural lung damage.5 Furthermore, the conventional anti-inflammatory agents including corticosteroids may be insufficient for treating severe asthma associated with fungal sensitisation.29 In this study, treatment with dexamethasone failed to alleviate various pathophysiologic features of Af-induced allergic lung inflammation, including eosinophil-dominant inflammatory cell infiltration into the lung, AHR, and elevation of TH2-dominant pro-inflammatory cytokines. In fact, we revealed that under an identical protocol to this study, the same dose of dexamethasone attenuates the typical features of allergic asthma induced by ovalbumin which is a well established classic asthma model.19 These results suggest that Af-induced lung inflammation in the current murine experimental model represents a unique endotype for steroid-resistant eosinophil-dominant allergic lung inflammation.
ER stress and UPR activation are known to be closely involved in pathogen immunity by influencing innate and adaptive immune responses and being interconnected with diverse intracellular inflammatory platforms.30 Previous studies have demonstrated that ER stress is implicated in the pathogenesis of various pulmonary disorders, including bronchial asthma, COPD, idiopathic pulmonary fibrosis and lipopolysaccharide-related acute lung injury.19 ,31–33 Particularly, we recently reported that ER stress contributes to corticosteroid resistance in a murine model of neutrophil-dominant allergic lung inflammation.19 In the current study, sensitisation/challenge with Af increased expression of ER stress markers and UPR-related proteins in the lung. Expression of ER stress markers was also elevated in Af-stimulated TECs. Moreover, in lung tissues from patients with ABPA, expression of GRP78 was also increased. In addition, blockade of ER stress improved Af-induced eosinophilic allergic lung inflammation and AHR. However, Af-induced increases in the levels of GRP78 and CHOP were not reduced by treatment with dexamethasone. These findings suggest that ER stress is involved in the pathogenesis of Af-induced allergic lung inflammation.
Activation of transcription factor NF-κB serves as an important molecular platform interconnecting ER stress and inflammatory process.17 Recently, the NF-κB signalling pathway has been reported to be essential in ER stress-related lung inflammation.19 ,33 In the present study, the level of nuclear NF-κB p65 in the lung was significantly increased after Af challenge. Af-induced increase in NF-κB p65 was decreased by administration of an ER stress inhibitor. Additionally, treatment with a NF-κB inhibitor substantially reduced increases in pro-inflammatory mediators, inflammatory cell infiltrations specifically eosinophils, and AHR in Af-exposed mice. Taken together, these findings suggest that NF-κB is a pivotal player in ER stress-related allergic lung inflammation.
The PI3K pathway mediates a number of critical cellular events, such as cell survival, proliferation and migration.7 As for Af-related inflammatory disorders, to date, it remains unclear about the roles of PI3K and its isotypes. In fact, the PI3K pathway has been known to participate not only in host defence against Af34 ,35 but also in the pathogenesis of Af-induced inflammatory disorders or aspergillosis.36 In addition, there is little available information on the PI3K-δ subtype in Af-related pulmonary disorders. Previous studies have reported that PI3K-δ is crucially involved in the pathogenesis of a number of pulmonary inflammatory diseases, including bronchial asthma and COPD.13 ,37 In particular, PI3K-δ is known to mediate steroid resistance in relation to neutrophil-dominant inflammatory processes in the lung. The basic mechanisms through which PI3K-δ modulates neutrophilic inflammation include trafficking and activation of neutrophils, and decrease in histone deacetylase-2 expression.11 ,13 ,15 However, severe allergic lung inflammation is heterogeneous and may arise from diverse pathophysiological processes involving various cell types. Among them, eosinophilic inflammation affects the majority of patients with severe asthma.38 In our study, Af challenge in mice increased levels of PIP3 and p-Akt in the lung. Stimulation with Af also upregulates PI3K-δ mRNA expression in TECs. In Af-exposed mice, PI3K-δ inhibition with a selective inhibitor significantly lowered Af-induced increases in PIP3 and p-Akt and reduced infiltration of airway inflammatory cells, especially eosinophils, production of pro-inflammatory mediators, total and Af-specific IgE, and AHR. Interestingly, PI3K-δ inhibition significantly limited Af-induced increases in ER stress markers and UPR-related proteins in the lung. In addition, the PI3K-δ inhibitor significantly limited Af-induced increases in ER stress markers in TECs. Transfection of PI3K-δ siRNA also significantly limited Af-induced increases in GRP78 and CHOP in cells. In agreement with these findings, blockade of PI3K-δ using the PI3K-δ inhibitor or PI3K-δ siRNA in cells substantially limited Af-induced increases in immunofluorescence intensities of GRP78 and CHOP. These observations indicate that PI3K-δ may play an important role in Af-induced steroid-resistant eosinophil-dominant allergic lung inflammation. Moreover, PI3K-δ seems to contribute to steroid resistance in the current model through induction of ER stress in the lung.
ROS production, which leads to oxidative stress, is one of critical features in chronic airway disorders.26 In addition, oxidative stress contributes to diverse pathologic processes through modulation of the PI3K signalling pathway.39 Emerging evidence has indicated that mitochondrion is an important source in the production of most intracellular ROS and that considerable interplay between mitochondria and ER in several respiratory disorders has been demonstrated.27 ,40 Moreover, PI3K signalling is closely associated with mitochondrial physiology.41 In the present study, BAL cells of Af-exposed mice and Af-stimulated TECs showed increased production of mtROS. In addition, blockade of PI3K-δ reduced Af-induced increases in mtROS generation in these cells. PI3K-δ inhibition also blocked Af-induced decreases in GSH level in the lung of Af-exposed mice. Moreover, treatment with NecroX-5 effectively attenuated increases in mtROS in BAL cells, reduced increases in GRP78 and CHOP in Af-exposed mice, and ameliorated pathophysiologic features of Af-induced allergic airway inflammation. Taken together, PI3K-δ may activate the ER stress pathway at least in part through regulation of mtROS generation in the current model.
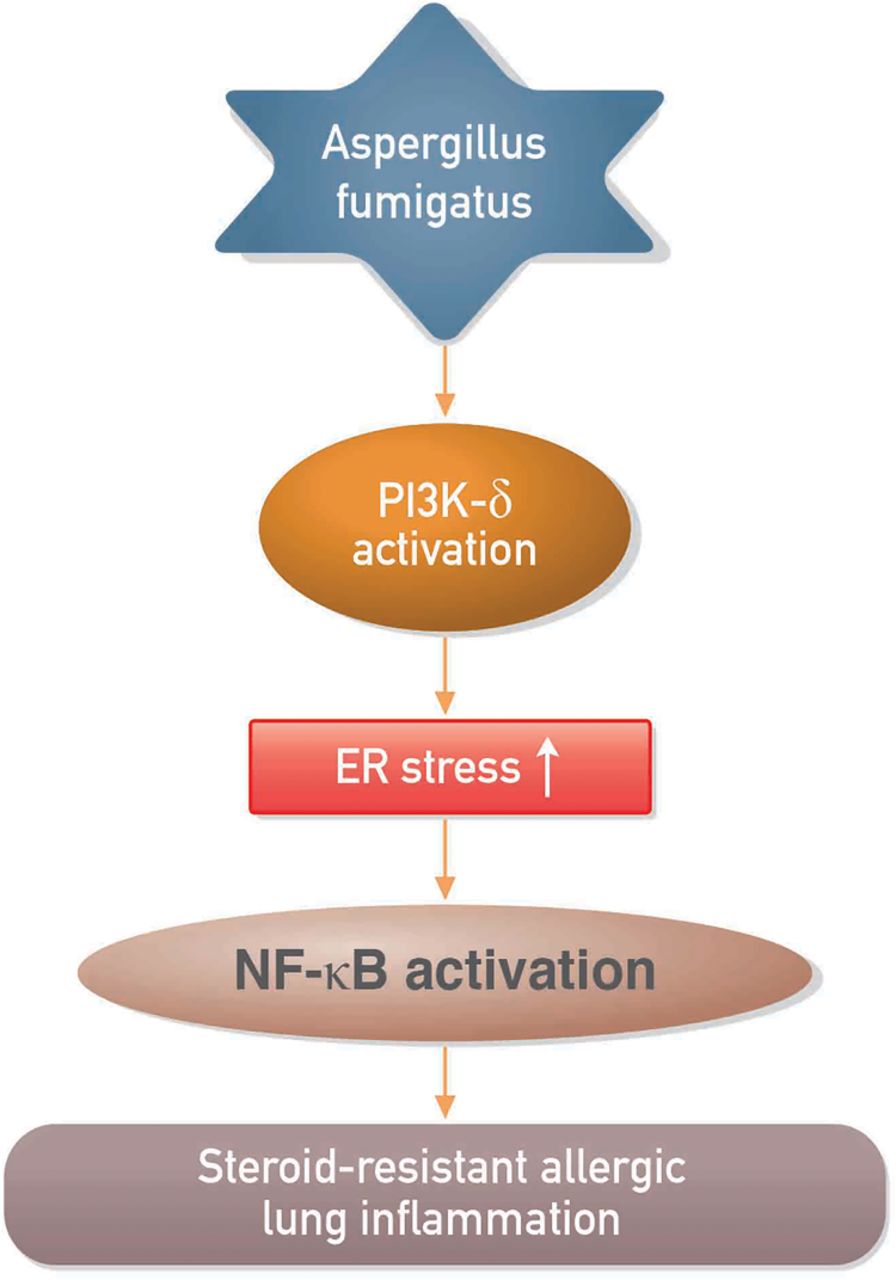
In summary, we have shown that PI3K-δ and ER stress are involved in the pathogenesis of Af-induced allergic lung inflammation and that PI3K-δ may regulate Af-induced steroid-resistant lung inflammation through ER stress (figure 8). Our results suggest that inhibition of PI3K-δ is efficacious in preclinical models of fungus-induced severe allergic lung disease and may have potential for treating patients with steroid-resistant eosinophil-dominant allergic lung disorders.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed mechanism for the involvement of phosphoinositide 3-kinase (PI3K)-δ in the regulation of endoplasmic reticulum (ER) stress in Aspergillus fumigatus (Af)-related steroid-resistant allergic lung inflammation. NF-κB, nuclear factor κB.
Acknowledgments
We thank Professor Mie-Jae Im and Peter Lee for critical readings of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
KSL, JSJ and SRK contributed equally.
Contributors KSL performed experiments and analysed data; JSJ interpreted data and wrote the manuscript; SRK designed research, interpreted data, and edited the manuscript; SHC reviewed and edited the manuscript; NK reviewed and edited the manuscript; YHK performed experiments and analysed data; KBL analysed data and reviewed the manuscript; SCP analysed data and reviewed the manuscript; HJP analysed data and reviewed the manuscript; YCL designed research, interpreted data, and edited the manuscript.
Funding This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2014R1A2A1A01002823).
Competing interests None declared.
Ethics approval Institutional Review Board of the Biomedical Research Institute of Chonbuk National University Hospital (IRB file No. 2013-11-007-001).
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves