Article Text

Abstract
Rationale Respiratory tract infections are common in patients suffering from pulmonary fibrosis. The interplay between bacterial infection and fibrosis is characterised poorly.
Objectives To assess the effect of Gram-positive bacterial infection on fibrosis exacerbation in mice.
Methods Fibrosis progression in response to Streptococcus pneumoniae was examined in two different mouse models of pulmonary fibrosis.
Measurements and main results We demonstrate that wild-type mice exposed to adenoviral vector delivery of active transforming growth factor-β1 (TGFß1) or diphteria toxin (DT) treatment of transgenic mice expressing the DT receptor (DTR) under control of the surfactant protein C (SPC) promoter (SPC-DTR) to induce pulmonary fibrosis developed progressive fibrosis following infection with Spn, without exhibiting impaired lung protective immunity against Spn. Antibiotic treatment abolished infection-induced fibrosis progression. The cytotoxin pneumolysin (Ply) of Spn caused this phenomenon in a TLR4-independent manner, as Spn lacking Ply (SpnΔply) failed to trigger progressive fibrogenesis, whereas purified recombinant Ply did. Progressive fibrogenesis was also observed in AdTGFβ1-exposed Ply-challenged TLR4 KO mice. Increased apoptotic cell death of alveolar epithelial cells along with an attenuated intrapulmonary release of antifibrogenic prostaglandin E2 was found to underlie progressive fibrogenesis in Ply-challenged AdTGFβ1-exposed mice. Importantly, vaccination of mice with the non-cytotoxic Ply derivative B (PdB) substantially attenuated Ply-induced progression of lung fibrosis in AdTGFβ1-exposed mice.
Conclusions Our data unravel a novel mechanism by which infection with Spn through Ply release induces progression of established lung fibrosis, which can be attenuated by protein-based vaccination of mice.
- Idiopathic pulmonary fibrosis
- Respiratory Infection
- Bacterial Infection
Statistics from Altmetric.com
Key messages
What is the key question?
Little knowledge exists concerning whether bacterial infections may cause exacerbation of pulmonary fibrosis.
What is the bottom line?
The study shows that lung tropic bacteria may trigger progressive lung fibrosis through release of defined cytotoxic virulence factors promoting alveolar epithelial cell apoptosis in mice.
Why read on?
The current study contributes to a better understanding of infection-induced lung fibrosis exacerbation using two different preclinical mouse models.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most severe interstitial lung disease with a prevalence of 2–29 cases per 100 000 individuals and a median survival ranging from 3 to 4 years.1 The disease process is characterised by repeated alveolar epithelial micro-injury resulting in epithelial cell damage, accumulation of fibroblasts and myofibroblasts along with increased extracellular matrix deposition.2 This leads to progressive tissue scarring and formation of honeycomb cysts, collectively causing irreversible loss of lung function and respiratory failure. Many patients with IPF experience unpredictable episodes of acute worsening of their disease. These can result from secondary complications, such as PE, bacterial and/or viral pneumonia, or for unknown reasons, which are then termed acute exacerbation of IPF (AE-IPF).3–5 While there is no direct evidence that viral infections are present in patients with AE-IPF,6 the clinical presentation is consistent with an infectious trigger of these events.7–9 Previous reports have demonstrated that γ-herpes virus can mediate exacerbation in lung fibrosis in mice.3 ,10 Most recent work on the role of bacteria in progression of IPF concluded that IPF progression is associated with the presence of specific members within the Streptococcus and Staphylococcus genera, which predicts decline in lung function and death.11 ,12 However, it is still unclear whether bacterial infections in IPF are merely a secondary complication in a high-risk patient group, or if they can also worsen the underlying fibrosis. This is a clinically highly relevant issue considering that patients with IPF may be more susceptible to develop bacterial pneumonias.8
In this study, we wanted (1) to examine if animals with experimental lung fibrosis are impaired in their ability to clear bacterial infections and (2) to analyse whether bacterial infections are able to trigger worsening of lung fibrosis. Two independent models of pulmonary fibrosis in mice were employed. The first model consists of adenoviral vector delivery of active transforming growth factor-β1 (TGFβ1) gene into the lungs of mice to induce pulmonary fibrosis.13 ,14 The other mouse model consists of diphtheria toxin (DT) administration to transgenic surfactant protein C-diphteria toxin receptor (SPC-DTR) mice to cause repetitive alveolar type II epithelial cell injury and subsequent pulmonary fibrosis.15 As a second hit, mice with established lung fibrosis underwent pulmonary infection with the prototypic community-acquired lung pathogen, Streptococcus pneumoniae (Spn).16
Methods
All animal experiments were approved by local government authorities. The methods are outlined in detail in the online supplementary data.
Results
Characterisation of AdTGFβ1-induced lung fibrosis in mice and its effect on lung antibacterial immunity
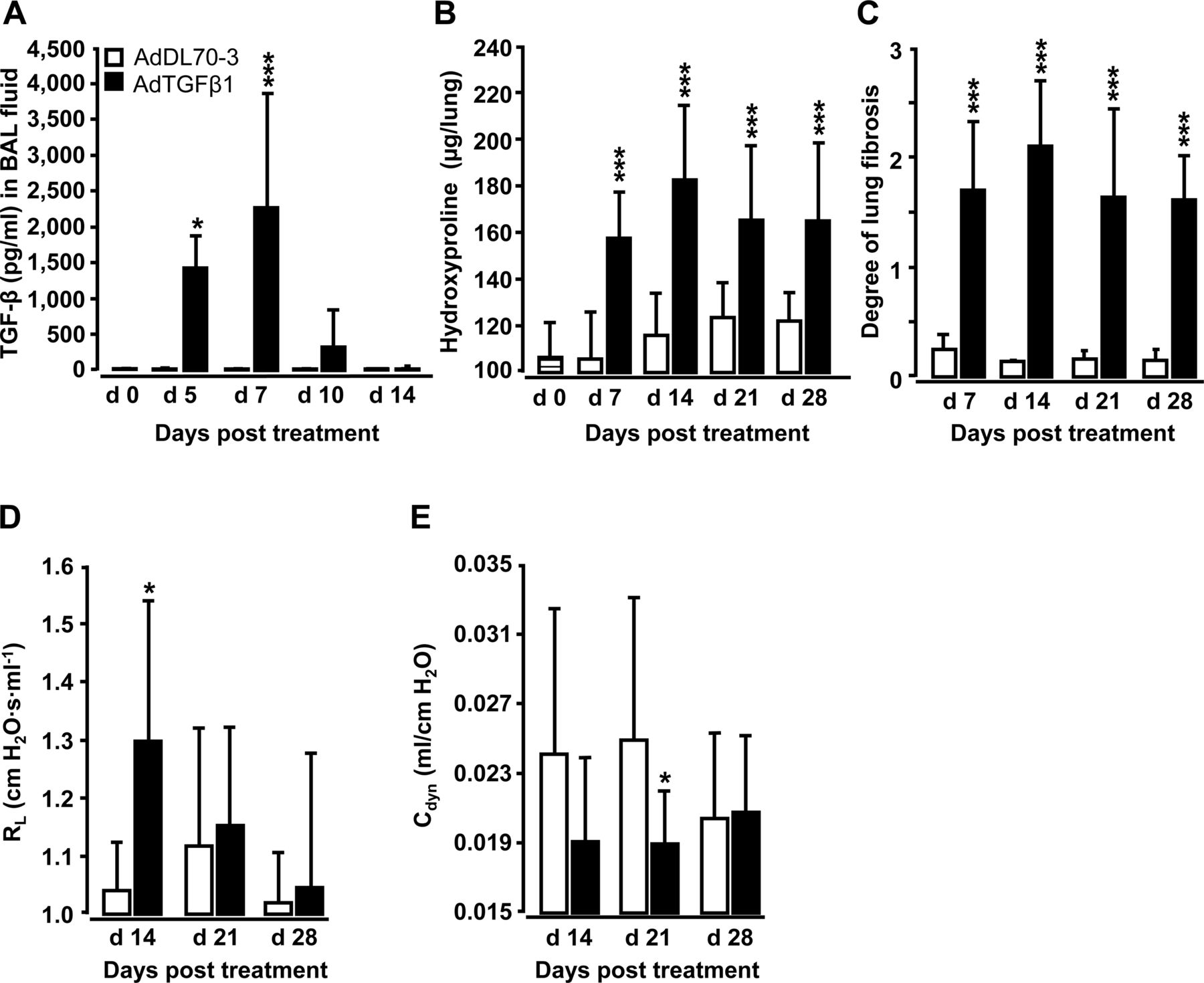
Orotracheal (o.t.) application of AdTGFβ1 resulted in increased protein levels of active TGFβ1 in bronchoalveolar lavage (BAL) fluid (BALF) by day 5, peaking by day 7 and declining thereafter (figure 1A). Increased TGFβ1 levels were followed by significantly increased hydroxyproline levels by day 7, peaking by day 14 and remaining increased until day 28 after AdTGFβ1 application (figure 1B). Morphometric assessment of lung fibrosis revealed significantly increased Ashcroft scores in AdTGFβ1-treated mice relative to controls (figure 1C). We also observed significantly increased lung resistance (RL) by day 14 and decreased dynamic compliance (Cdyn) by day 21 post-AdTGFβ1, relative to controls (figure 1D, E).
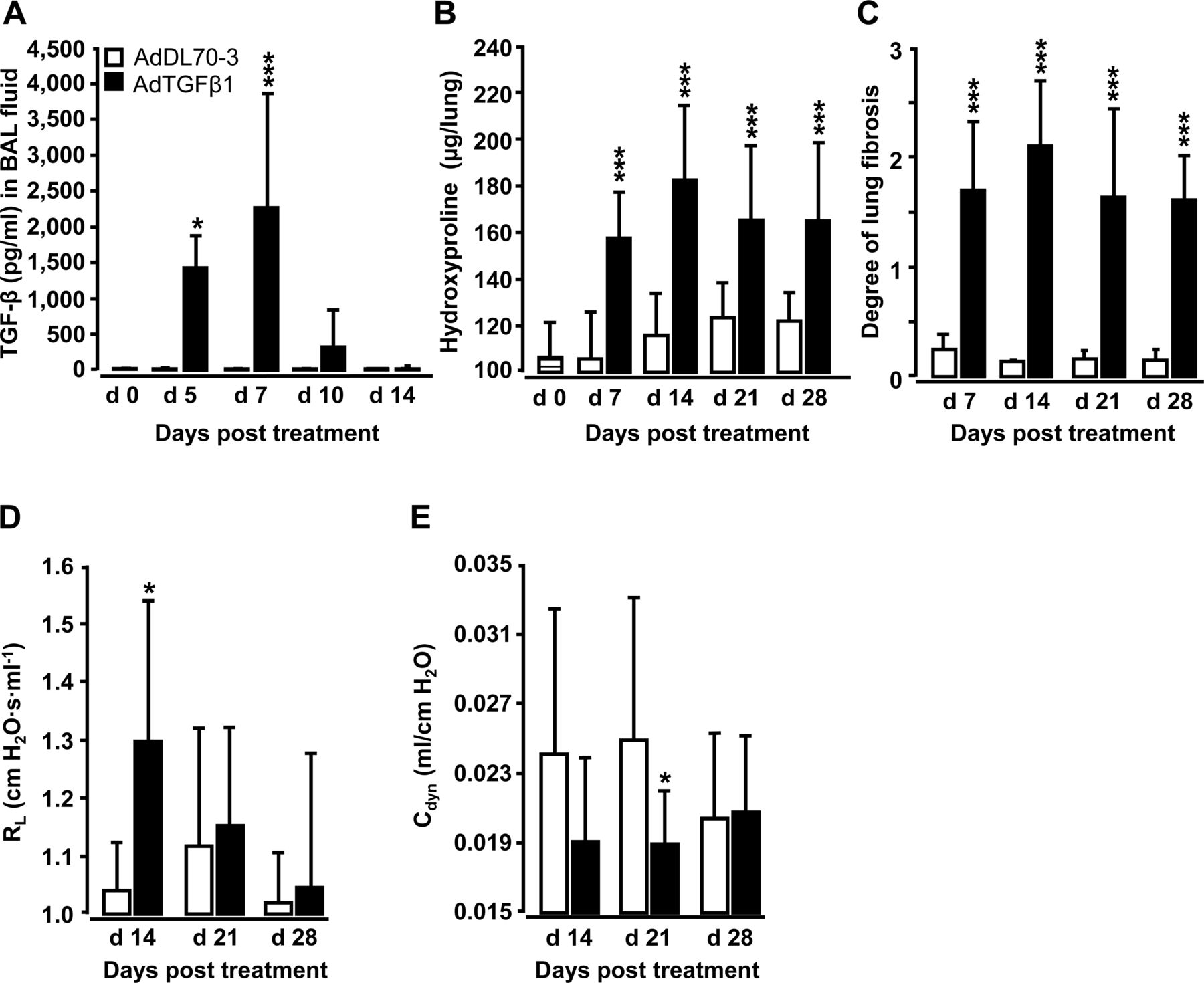
Characterisation of AdTGFβ1-induced lung fibrosis in mice. Naïve mice (lined bars, day 0 time points) were treated with control vector AdDL70-3 (1×108 PFU, white bars) or AdTGFβ1 (1×108 PFU, black bars) for various time points, as indicated. (A) Kinetics of active TGFβ1 protein in BAL fluid (BALF). (B) Analysis of hydroxyproline as a measure of lung collagen content in lung tissue. (C) Degree of lung fibrosis determined by histomorphometry/Ashcroft scoring. (D and E) Lung function in AdTGFβ1 and control-vector-exposed mice assessed by resistance (RL, D) and dynamic compliance (Cdyn, E). Data are shown as mean±SD of n=5 mice per time point and treatment group (n=10 mice per treatment group in D and E) and are representative of three (A–C) or two independently performed experiments (D and E). Data were analysed by Student's t test. *p<0.05, ***p<0.001 compared with control-vector-treated mice. PFU, plaque-forming unit.
Next, we examined the lung host defence of mice undergoing AdTGFβ1 treatment for 14, 21 or 28 days, followed by S. pneumoniae (Spn) infection. No significant differences in bacterial loads were observed between groups of Spn-infected AdTGFβ1 or control-vector-treated mice (figure 2A–C). Moreover, no significant differences in numbers of BALF cells or levels of proinflammatory TNF-α were detected between groups of mice exposed for 14 days to AdTGFβ1 versus control vector followed by Spn infection (figure 2D–F).

Effect of AdTGFβ1 on lung protective immunity against bacterial challenge. (A–C) Bacterial loads were determined in lung tissue at 24, 48 and 72 h after Spn infection (1×107 CFU/mouse) from mice exposed to AdTGFβ1 (black bars) or control vector (white bars) for 14 days (A), 21 days (B) or 28 days (C), as indicated. (D–F) BAL fluid (BALF) was collected from untreated control mice (striped bars) or at 24, 48 and 72 h after Spn infection from mice exposed to AdTGFβ1 or control vector for 14 days and numbers of macrophages (D) and neutrophils (E) as well as TNF-α protein levels (F) were determined. Data are shown as mean±SD of n=4 mice per time point and treatment group, and are representative of three independently performed experiments. Data were analysed by Mann–Whitney U test. CL, untreated control mice; CFU, colony-forming unit.
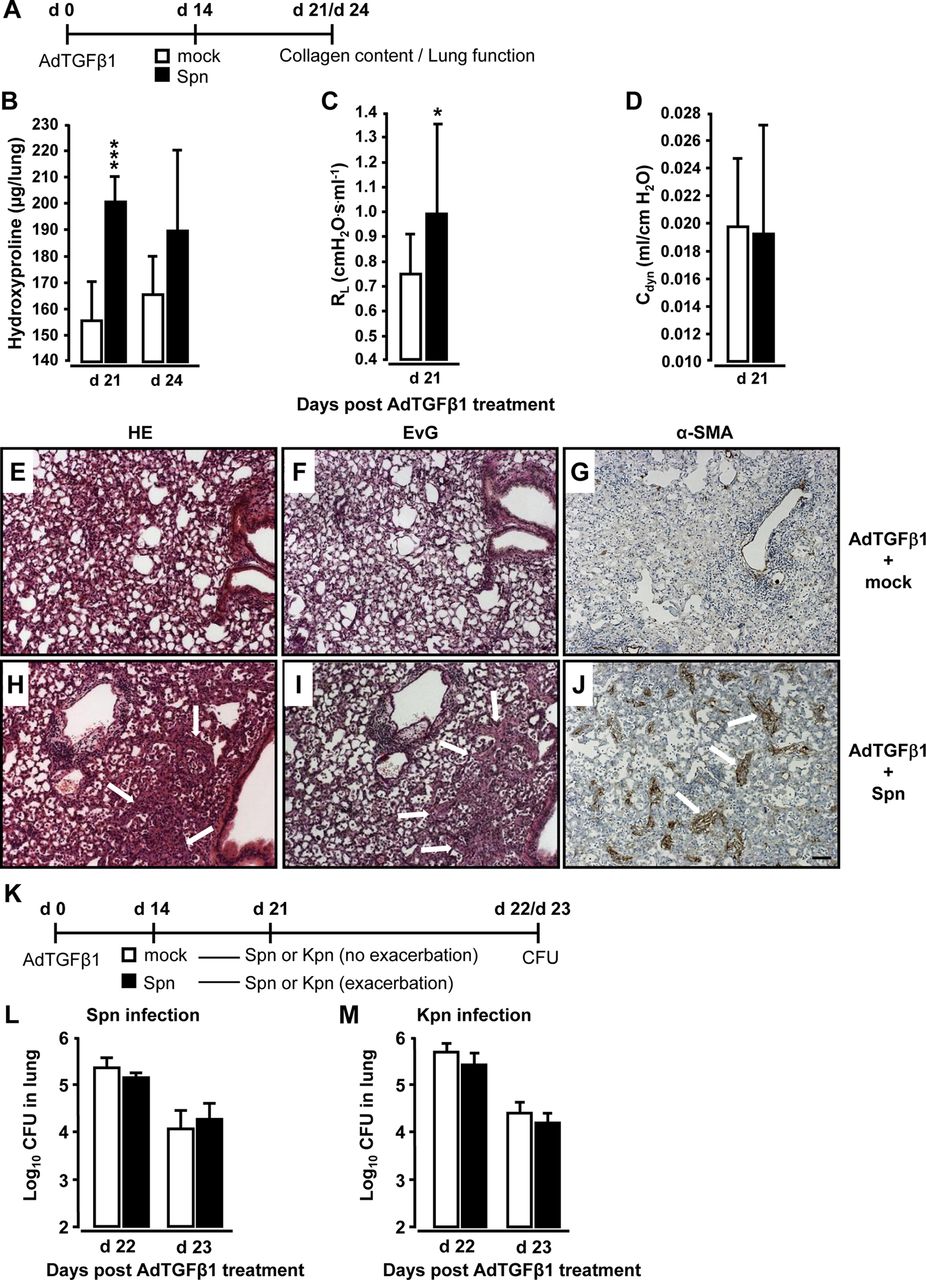
Infection with S. pneumoniae triggers exacerbation of pulmonary fibrosis in mice
Mice were exposed to AdTGFβ1 and 14 days later were infected with Spn or mock for another 7 or 10 days (figure 3A). Significantly increased collagen deposition was noted on days 21 and 24 post-AdTGFβ1 in mice infected with Spn for 7 days (figure 3B). Functional analysis on day 21 revealed significantly increased lung resistance in Spn- relative to mock-infected mice (figure 3C, D). Histopathological examination of lung tissue sections revealed that mice exposed to AdTGFβ1 followed by Spn infection showed substantially more bronchial and interstitial inflammation, fibrosis and alveolar epithelial hyperplasia on day 21 post-treatment relative to mock-infected mice (figure 3E, H). Marked thickening of the alveolar septa due to activated (myo-) fibroblasts (compare α-smooth muscle actin (SMA) stains in figure 3G, J) and increased collagen deposition (compare Elastica-van Gieson (EvG) stains in figure 3F, I) accompanied by prominent pneumocyte activation was observed in AdTGFβ1 plus Spn, but not mock-infected mice.

Bacterial infection triggers progressive pulmonary fibrosis in AdTGFβ1-exposed mice. (A) Experimental profile. Mice were exposed to AdTGFβ1 (1×108 PFU) for 14 days and were then either mock-infected (white bars) or Spn-infected (1×107 CFU, black bars) for 7 or 10 days, as indicated. (B) Lung collagen content in Spn-infected mice at day 7 and 10 post-Spn challenge. (C and D) Assessment of resistance (RL, C) and dynamic compliance (Cdyn, D) in the lungs of AdTGFβ1-treated, Spn-infected mice on day 21 post-AdTGFβ1 (corresponding to day 7 post-Spn). (E–J) Histopathology of lungs from AdTGFβ1-treated mice subsequently challenged with mock (E–G) or Spn (H–J) for 7 days. Lung tissue sections were stained with H&E (E and H), Elastica-van-Gieson (EvG; F and I), and α-smooth muscle actin (α-SMA; G and J). Note substantially increased collagen deposition and myofibroblast accumulation in H, I and J (marked by white arrows). (K) Experimental profile to determine lung protective immunity of mice exhibiting Spn-induced progression of AdTGFβ1-induced lung fibrosis. On day 14 post-AdTGFβ1 (1×108 PFU), mice were either mock-infected (white bars) or Spn-infected (1×107 CFU, black bars) for 7 days, resulting in fibrosis progression. Subsequently, mice were reinfected with Spn (1×107 CFU, L) or Kpn (5×106 CFU, M) on day 7 after initial mock or primary bacterial infection, and bacterial loads were determined, as indicated (L and M). The data are shown as mean±SD of n=5 mice per time point and treatment group. The experiment was repeated twice with similar results. Data were analysed by Student's t test (B–D) and Mann–Whitney U test (L and M). *p<0.05, ***p<0.001 compared with mock-treated mice. (E–J) Scale bar, 100 µm. CFU, colony-forming unit; PFU, plaque-forming unit.
We further examined whether Spn-induced worsening of pulmonary fibrosis might impair lung protective immunity against bacterial infection. By day 14 post-AdTGFβ1, mice were infected with Spn or mock for 7 days and received another challenge with either Spn or Klebsiella pneumoniae (Kpn) (Kpn to exclude immunological effects due to the previous Spn infection, see figure 3K). Importantly, AdTGFβ1-exposed mice developing progressive lung fibrosis in response to the first Spn challenge did not show any defect in bacterial killing after the second Spn or Kpn infection (figure 3L, M).
Bacterial infection triggers progressive lung fibrosis in a model of DT-induced pulmonary fibrosis
As an alternative model of pulmonary fibrosis, we employed transgenic SPC-DTR mice expressing the human DT receptor on AT II cells and responding to DT administration with pulmonary fibrosis.15 Orotracheal application of DT for seven consecutive days induced pulmonary fibrosis in transgenic SPC-DTR mice, but not in vehicle- or DT-treated wild-type (WT) mice at day 14 post-application (figure 4A, B). Challenge of transgenic SPC-DTR mice with Spn at day 14 post-DT treatment also triggered progressive lung fibrosis in this model, while WT littermates did not respond with progressive fibrosis to DT and Spn treatment (figure 4C, D).

Bacterial infection triggers progressive pulmonary fibrosis in diphtheria toxin (DT)-exposed transgenic SPC-DTR mice. (A and B) Characterisation of DT-induced lung fibrosis in transgenic SPC-DTR mice. Transgenic SPC-DTR mice and their wild-type (WT) littermates were either treated o.t. with vehicle (PBS) or DT daily for 7 days. (B) Lung collagen content in WT and transgenic SPC-DTR mice at day 14 after DT treatment. (C) Experimental profile. SPC-DTR mice and their WT littermates received daily o.t. applications of DT for 7 days and were then challenged with mock versus Spn on day 14, that is, the time of established lung fibrosis. (D) Lung collagen content in WT and SPC-DTR mice treated with DT followed by mock infection or Spn infection, as indicated in (C). The data are shown as mean±SD of n=5 mice per time point and treatment group, and is representative of two independently performed experiments. Data were analysed by two-way ANOVA. **p<0.01, compared with vehicle-treated SPC-DTR mice (B), ***p<0.001 compared with DT-treated WT mice, +p<0.05 compared with mock-infected SPC-DTR mice (D). ANOVA, analysis of variance; o.t., orotrachea; PBS, phosphate-buffered saline.
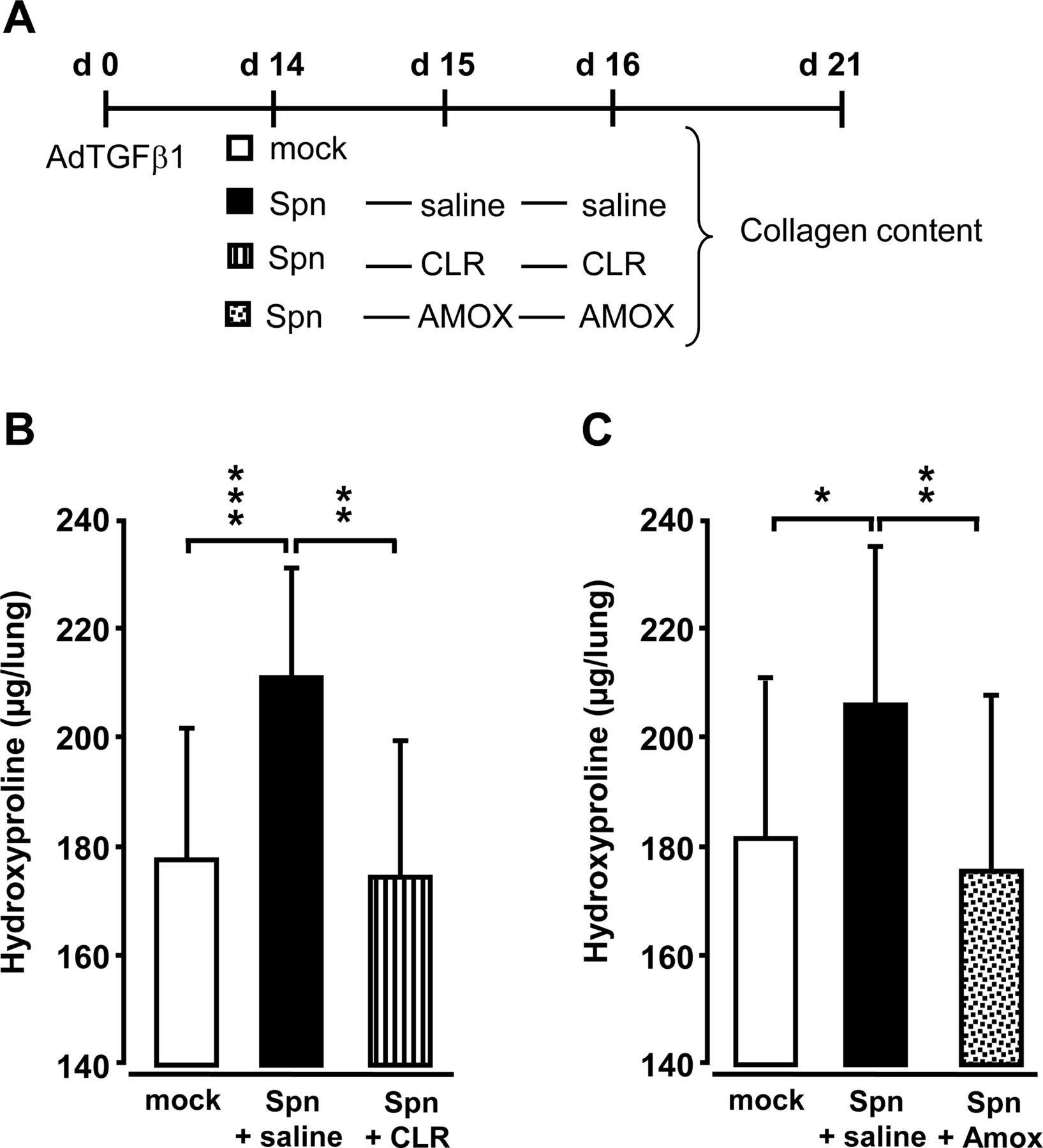
Effect of antibiotic treatment on Spn-induced progression of lung fibrosis in mice
At day 14 post-AdTGFβ1, mice were either Spn- or mock-infected followed by either saline, or clarithromycin or amoxicillin treatment (figure 5A). Importantly, antibiotic treatment with clarithromycin or amoxicillin at 24 h and 48 h post-infection led to significantly decreased lung hydroxyproline contents, comparable with mock-infected mice (figure 5B, C).

Effect of antibiotic treatment on Spn-induced exacerbation of lung fibrosis in mice. (A) Experimental profile. Mice were pretreated with AdTGFβ1 (1×108 PFU/mouse) for 14 days and were then either mock-infected (white bars) or Spn-infected (1×107 CFU/mouse). Subsequently, mice received saline (black bars), clarithromycin (CLR, 50 mg/kg b.w., striped bars) or amoxicillin (AMOX, 100 mg/kg b.w., dotted bars) on day 1 and 2 post-infection (corresponding to days 15 and 16 post-AdTGFβ1). (B and C) Lung collagen contents in the lungs of mice of the respective treatment groups determined on day 21 post-AdTGFβ1. The data are shown as mean±SD of n=3 mice per time point and treatment group, and is representative of four independently performed experiments. Data were analysed by one-way ANOVA. *p<0.05, **p<0.01, ***p<0.001 compared with Spn-infected + saline-treated mice. ANOVA, analysis of variance; b.w., body weight; CFU, colony-forming unit; PFU, plaque-forming unit.
Role of pneumolysin in Spn-induced progression of lung fibrosis
Spn is able to cause alveolar epithelial injury in murine lungs by release of the pore-forming cytotoxin pneumolysin (Ply).17 At the same time, Ply is a bacterial PAMP and is known to induce Toll-like receptor 4 (TLR4) signalling in lung effector cells such as macrophages.18 Therefore, we hypothesised that Spn-derived Ply might cause fibrosis exacerbation in Spn-infected mice. Challenge of AdTGFβ1-exposed mice with a Ply-deficient isogenic mutant of Spn, SpnΔply, did not cause fibrosis exacerbation (figure 6A, B). Similarly, challenge of SPC-DTR mice with established lung fibrosis with Ply-deficient Spn also failed to induce fibrosis exacerbation, just opposite to Spn challenge of DT-pretreated SPC-DTR mice, which responded to Spn infection with significantly increased hydroxyproline levels (figure 6C, D).

Effect of pneumolysin on progression of AdTGFβ1 and SPC-DTR-dependent lung fibrosis. (A) Experimental profile. Mice were exposed to AdTGFβ1 (1×108 PFU/mouse) for 14 days and were then either mock-infected (white bars) or Spn-infected (1×107 CFU/mouse, black bars) or were infected with the isogenic mutant strain of Streptococcus pneumoniae SpnΔply (1×107 CFU/mouse, grey bars). (B) On day 21, lungs were harvested and lung hydroxyproline contents were determined. (C) Experimental profile. SPC-DTR transgenic mice received daily DT applications for 7 days orotracheally and were then mock-infected (white bars) or Spn-infected (1×107 CFU/mouse, black bars) or were infected with the isogenic mutant strain of S. pneumoniae SpnΔply (1×107 CFU/mouse, grey bars) on day 14. (D) On day 21, lungs were harvested and lung hydroxyproline contents were determined. (E) Lung permeability determined in naïve wild-type (WT) mice (white bar) or mice at 6 h after single challenge with Ply (black bars), as indicated. (F) Experimental profile for repetitive treatment of AdTGFβ1-exposed WT mice with Ply. Mice were exposed to AdTGFβ1 (1×108 PFU/mouse) for 14 days, and on days 14, 15 and 16, mice received PBS o.t. (50 µL, white bars) or either PdB or PdT or Ply (∼50 haemolytic units per mouse) in 50 µL PBS (black bars), as indicated. (G) On day 21, lungs were harvested for determination of collagen contents. (H) Experimental profile. WT and TLR4 KO mice were exposed to AdTGFβ1 (108 PFU/mouse) for 14 days, and on days 14, 15 and 16, mice received PdB (white bars) or Ply (∼50 haemolytic units per mouse, black bars) o.t. for three consecutive days. (I) On day 21, lungs were harvested for determination of collagen contents. The data are shown as mean±SD of n=5 mice per time point and treatment group and are representative of two independently performed experiments. Data were analysed by one-way ANOVA (B, D, G) and two-way ANOVA (I). *p<0.05, **p<0.01 compared with mock and SpnΔply treatment in (B, D), and compared with mock or PdB or PdT treatment in (G), and compared with PdB treatment (I). ANOVA, analysis of variance; CFU, colony-forming unit; o.t., orotracheal; PBS, phosphate-buffered saline; PdB, pneumolysin derivative B; PdT, pneumolysin derivative T; PFU, plaque-forming unit.
To confirm the pivotal role of Ply in triggering fibrosis progression, we next titrated Ply to induce epithelial injury without causing mortality (figure 6E). Application of 100 ng Ply/mouse lung triggered increased lung permeability at 6 h post-treatment without any mortality, whereas 125 and 150 ng Ply/mouse lung caused profound lung permeability with higher mortality within 6 h (125 vs 150 ng, Ply: ∼50% vs 65% mortality). Based on these observations, we challenged mice with established lung fibrosis with 115 ng Ply (∼ 50 haemolytic units per mouse), or its non-haemolytic derivative pneumolysin derivative B (PdB) or its non-haemolytic non-complement activating derivative pneumolysin derivative T (PdT) once daily for three consecutive days (figure 6F). Application of Ply, but not PdB, or PdT triggered progression of AdTGFβ1-induced lung fibrosis in mice (figure 6G).
To examine whether Ply would trigger fibrosis progression through TLR4,18 we subjected AdTGFβ1-exposed WT and TLR4 knockout mice to repetitive Ply challenge (figure 6H). Importantly, AdTGFβ1-exposed TLR4 KO mice responded with a significantly increased fibrotic response to repetitive Ply challenge as WT mice, demonstrating that Ply-induced fibrosis progression is TLR4 independent and is based on the cytotoxic activity of the toxin (figure 6I).
Repetitive Ply challenge triggers apoptotic cell death in AT II cells
Repeated alveolar epithelial injuries may underlie the pathogenesis of human IPF.19 Based on our observation that Ply triggered fibrosis progression in mice via TLR4-independent pathways, we hypothesised that it might act via its pore-forming function to cause progressive epithelial injury in mice with established fibrosis, thereby exacerbating fibrogenesis. Therefore, we assessed the degree of early apoptosis induction in sorted AT II cells collected from the lungs of AdTGFβ1 plus Ply-challenged mice. FACS sorting of MHC IIpos AT II cells of AdTGFβ1 plus Ply-challenged mice resulted in sort purities of more than 98% (figure 7A–D). Indeed, AT II cells of AdTGFβ1-exposed mice responded to repetitive Ply challenge with significantly increased apoptosis assessed by annexin V staining in the absence of propidium iodide staining (figure 7E).
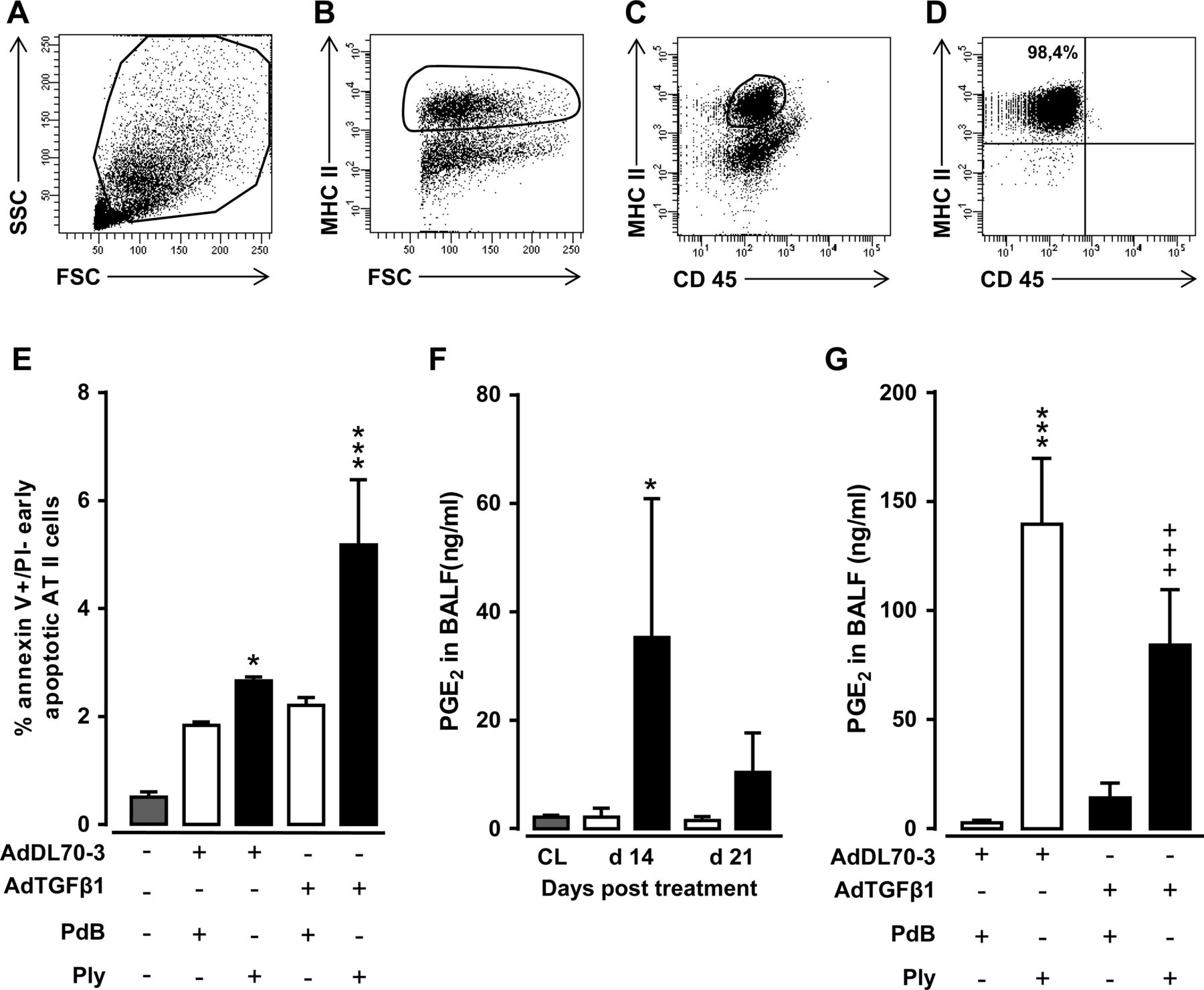
Effect of repetitive Ply challenge of AdTGFβ1-exposed mice on apoptosis and PGE2 levels of AT II cells. (A–D) Gating strategy for flow sorting of AT II cells isolated on day 21 from the lungs of mice exposed to AdTGFβ1 for 14 days followed by repetitive PdB or Ply challenge, as outlined in the legend to figure 6F. (A–D) Pre-enriched AT II cells were stained with anti-MHCII and anti-CD45 antibodies in the presence of anti-CD16/32 Ab, and were then gated according to their FSC-A versus SSC-A characteristics (A), followed by hierarchical subgating according to their FSC versus MHC II (B) and CD45 versus MHC II (C) expression. (D) Post-sort analysis of sorted AT II cells revealed sort purities of >98%, as verified by modified Papanicolaou stain and electron microscopic detection of lamellar bodies (data not shown). (E) Percent of early apoptotic (annexin Vpos, propidium iodideneg) AT II cells purified from the lungs of untreated mice (grey bars), or mice challenged with control vector (white bars) or AdTGFß1 (black bars) for 14 days and PdB or Ply for 7 days, as outlined in the legend to (E). (F) PGE2 levels in BAL fluids of untreated mice (grey bars) or mice treated with control vector (white bars) or AdTGFβ1 (black bars) for the indicated time intervals. (G) PGE2 levels in BAL fluids of mice treated with control vector or AdTGFβ1 for 14 days, followed by repetitive treatment with PdB versus Ply on days 14, 15 and 16 post-vector treatment, as indicated. PGE2 levels were determined on day 17 after vector treatment. The data are shown as mean±SD of n=3 mice per time point and treatment group (n=5 mice per time point and treatment group in F), and are representative of two independently performed experiments. Data were analysed by two-way ANOVA (E and G) or Student's t test (F). *p<0.05, ***p<0.001 compared with control vector, +PdB treatment and +++p<0.001 compared with control vector +Ply treatment. ANOVA, analysis of variance; FSC, forward scatter; PdB, pneumolysin derivative B.
We next questioned how the observed Ply-induced increased apoptosis induction in AT II cells of AdTGFβ1-exposed mice might impact on lung fibrosis progression. Several reports have established an important role for epithelial cell-derived arachidonic acid metabolite prostaglandin E2 (PGE2) to regulate both lung fibroblast and myofibroblast proliferation as well as extracellular matrix deposition by its antifibrogenic effect on serum response factor-dependent extracellular matrix transcription.20–22 Therefore, we examined what impact repetitive Ply challenge would have on the capability of fibrotic lungs to mount PGE2 responses. As shown in figure 7F, AdTGFβ1 exposure resulted in significantly increased PGE2 release in BALFs of mice at day 14–21 post-treatment. Repetitive Ply challenge of AdTGFβ1-exposed mice resulted in highly significant attenuated BALF PGE2 levels compared with Ply-treated control-vector-exposed mice (figure 7G), suggesting that mice with established lung fibrosis have lost their capability to mount appropriate antifibrotic responses to secondary Ply challenge.
Effect of pneumococcal-protein-based vaccination on Ply-induced fibrosis progression
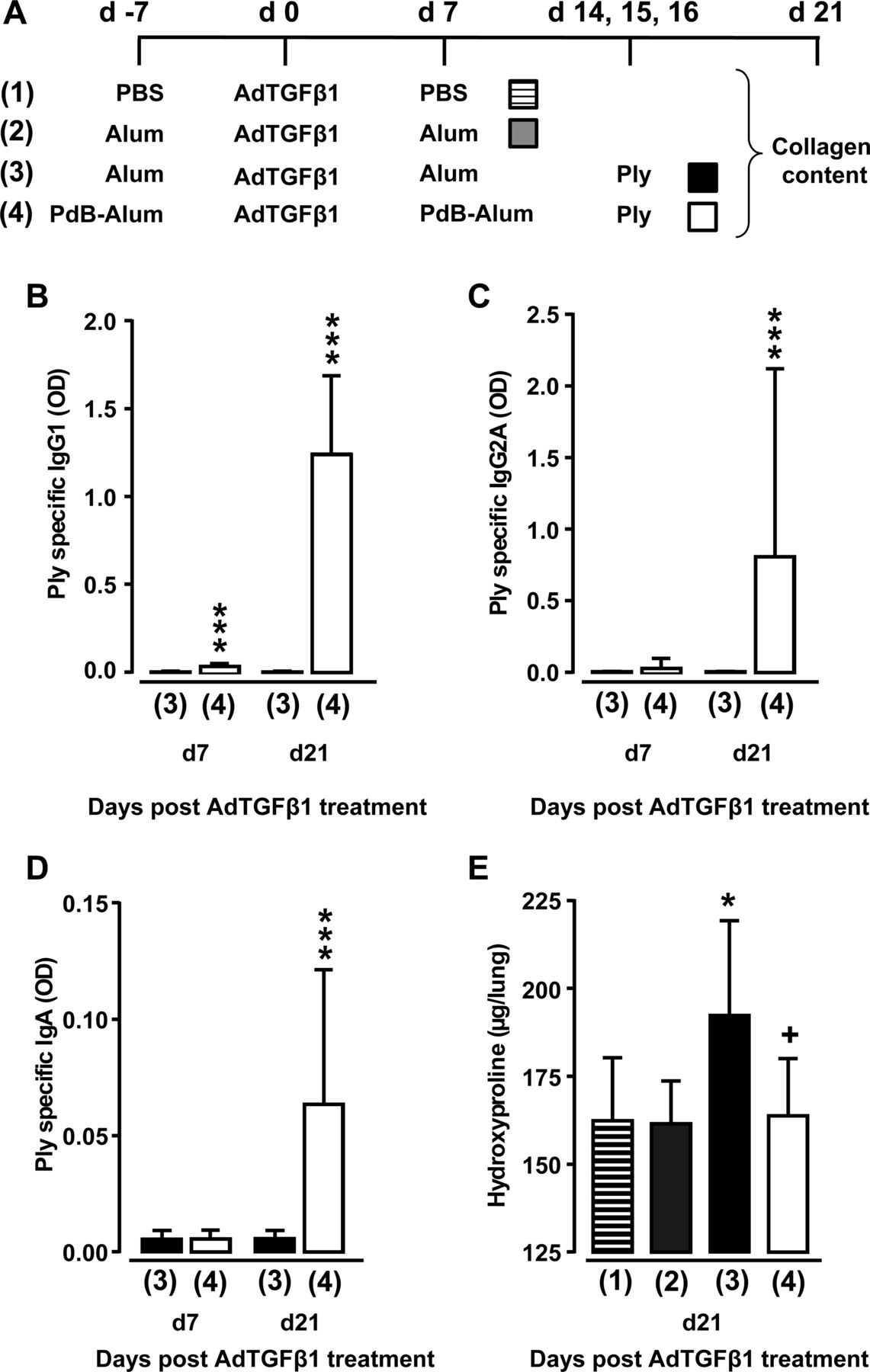
Having shown that Ply is a critical virulence factor of S. pneumoniae to cause progression of established lung fibrosis in two independent fibrosis models, we next examined whether protein-based vaccination would be a preventive strategy to inhibit Ply-induced fibrosis exacerbation in mice. As an immunogen, we chose the non-cytotoxic Ply derivative PdB, which was applied to mice according to the immunisation regimen outlined in figure 8A. As shown in figure 8B–D, primary and secondary immunisation of AdTGFβ1-exposed mice with PdB-Alum yielded significantly increased Ply-specific IgG1 and IgA antibody titres in plasma at 14 and 28 days post-primary vaccination (ie, 7 and 21 days post-AdTGFβ1 treatment), relative to adjuvans (Alum) in mice with established lung fibrosis. Importantly, PdB-vaccinated mice with established lung fibrosis did not respond to repetitive Ply challenge with fibrosis exacerbation when compared with Alum-treated Ply-challenged fibrotic mice (figure 8E). These data show that pneumococcal protein-based vaccination targeting critical pneumococcal virulence factors may have a major preventive effect on Spn-induced fibrosis exacerbation in mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of vaccination on Ply-induced fibrosis progression. (A) Experimental profile. On day -7, mice received PBS or Alum or PdB-Alum (intraperitoneal, 10 µg/mouse). On day 0, mice were exposed to AdTGFβ1, followed by re-immunisation with PdB-Alum (20 µg/mouse) or i.p. injection of Alum only or PBS on day 7 post-vector treatment, as indicated. On days 14, 15 and 16 after AdTGFβ1 exposure, mice received either PdB or Ply o.t., as indicated. (B–D) Plasma antibody titres of Ply specific IgG1 (B; 1:3000 diluted) or IgG2A (C; 1:30 diluted) or IgA (D; 1:30 diluted) from groups of mice as outlined in (A). (E) Lung hydroxyproline levels in experimental groups of mice as outlined in (A). The data are shown as mean±SD of n=9 mice per time point and treatment group, and are representative of three independently performed experiments. Data were analysed by Mann–Whitney U test (B–D) or one-way ANOVA (E). *p<0.05 compared with group 2, ***p<0.001 compared with group 3, +p<0.05 compared with group 3. ANOVA, analysis of variance; o.t., orotrachea; PBS, phosphate-buffered saline; PdB, pneumolysin derivative B.
Discussion
The clinical impression suggests that patients with IPF are more prone to bacterial respiratory tract infections. Pneumonias are among the most common reasons for death in IPF.1 ,23 However, there is no mechanistic evidence to explain this clinical observation. Here, we show that mice with established lung fibrosis were equally able to purge bacterial loads in distal lung airspaces compared with control mice, along with a similar extent of lung neutrophil and macrophage recruitment as well as similar levels of TNF-α release. These data suggest that the severity of lung fibrosis itself does not impact on lung protective immunity against bacteria. Even in more severe experimental fibrosis due to Spn-induced fibrosis exacerbation, we did not see an adverse effect on lung protective immunity against secondary bacterial infection.
We focused on elucidating the effect of bacterial infection on fibrosis exacerbation at 1 week post-infection, assuming that development of infection-induced fibrosis exacerbation would need some time. Therefore, we employed a serotype 19 S. pneumoniae in our experiments, which is known to primarily cause focal pneumonia. Use of more virulent pneumococcal strains rapidly causing invasive pneumococcal disease progression in immunocompetent mice was precluded in these experiments, as the resulting severe infection would have caused high mortality within a few days and terminated the experiments prior to the required endpoints.
Our study also demonstrates therapeutic efficacy of antibiotics in blocking Spn-induced fibrosis exacerbation in mice. Since both clarithromycin and amoxicillin provided similar efficacy in attenuating Spn-induced fibrosis exacerbation, possible immunomodulatory effects attributed to macrolide antibiotics24 ,25 may be largely excluded. This finding is of major clinical importance because patients with AE-IPF would most likely benefit from standardised antibiotic treatment regimen. Current practice for treating AE-IPF however is focused on immunosuppressive high-dose corticosteroids according to the recommendation in the official ATS/ERS/JRS/ALAT statement,1 but not on antibacterial drugs. Based on our and other data,26 administration of antibiotics to patients with COPD and IPF, once these patients have acquired either a viral and/or bacterial infection, should be considered with the intention to support anti-bacterial host defence and to prevent accelerated fibrogenesis and worsening of the underlying chronic lung disease. This is further supported by a recent study reporting that patients with IPF treated with twice-daily co-trimoxazole (960 mg) had significantly fewer events of rapid deterioration as long as they were treatment adherent.27 Considering that Ply is a cell-associated virulence factor released by S. pneumoniae upon cell lysis or death, and further given that Ply is shown here to cause fibrosis exacerbation, non-lytic antibiotics would be the preferred drug to inhibit infection-induced fibrosis exacerbation in patients with IPF.
Using two independent mouse models of pulmonary fibrosis, we here demonstrate for the first time that Spn infection of mice with established pulmonary fibrosis can cause substantial fibrosis progression, which could clinically resemble an episode of acute exacerbation. Infection of mice with a Ply-deficient isogenic mutant of Spn did not cause fibrosis exacerbation in both mouse models of lung fibrosis whereas repetitive application of recombinant purified Ply did. Less inflammation due to infection with a pneumolysin-deficient strain of Spn was also reported in earlier studies.28 ,29 Since the reported fibrosis exacerbation induced by Ply treatment was TLR4 independent, the findings support a role for Ply to act via its cytotoxic rather than its pathogen-associated molecular pattern (PAMP) signalling properties.
Increasing evidence supports a role for AT II cells in the pathogenesis of pulmonary fibrosis. We have applied Ply on three consecutive days to mimic the repetitive alveolar epithelial micro-injury, which is thought to contribute to the pathogenesis of IPF in humans.30 Using this modelling approach, repetitive Ply challenge increased early apoptosis in AT II cells of AdTGFβ-exposed mice. We and others have shown previously that Ply may exert toxic effects to alveolar epithelial cells due to its pore-forming activity.17 ,31 One possible explanation for the observed exacerbation-inducing effects of Ply in the current fibrosis models may be related to Ply-induced disturbance of prostaglandin-dependent crosstalk between AT II cells and fibroblasts. AT II cells play a role in inhibition of fibroblast proliferation and extracellular matrix deposition due to their ability to produce PGE2 32 Importantly, Ply-induced exacerbation of lung fibrosis was characterised by substantially decreased BALF PGE2 levels, relative to Ply-treated control-vector-exposed mice. These data support the view that opposed to a normal lung, a fibrotic lung has a substantially reduced capability to mount anti-fibrogenic responses after repetitive Ply-induced epithelial injuries probably due to cytotoxin-induced increased epithelial cell apoptosis. Since lung neutrophil recruitment was not impaired in mice with fibrosis exacerbation, the previously reported Ply-dependent PGE2 release by neutrophils is unlikely to account for the observed attenuated PGE2 levels in the fibrotic mice.33
Finally, we show for the first time that protein-based vaccination of mice with established lung fibrosis with the non-cytotoxic Ply-derivative PdB-protected mice from Ply-induced fibrosis exacerbation. We employed PdB as proteinaceous immunogen34 to mount Ply specific mucosa antibody responses in mice with established lung fibrosis to evaluate the importance of such a preventive strategy on Ply-induced fibrosis progression. The chosen protein-based vaccination approach has the major advantage of mounting mucosal immune responses in immunised hosts, which are characterised by antigen-specific secretory IgA responses, thereby mounting protection against cytotoxins such as Ply with a known tropism for epithelial cells as part of the lung mucosal surface. Transient breakdown of the epithelial barrier induced by pore-forming Ply may also facilitate access of Ply-specific antibodies to the alveolar lumen, thereby enabling neutralisation of intra-alveolar released Ply. The data of the current study strongly support the importance of vaccination to minimise the risk of infection-induced exacerbations in patients with chronic lung diseases such as pulmonary fibrosis.
Recently, it was suggested that the Gram-negative bacterial pathogen, Pseudomonas aeruginosa, as opposed to γ-herpes virus was not able to cause exacerbation in pulmonary fibrosis in mice.10 Several aspects need to be considered when comparing that study with our findings. First, as opposed to S. pneumoniae, P. aeruginosa is not a typical community-acquired pathogen. Since most of the patients with IPF are not hospitalised at the time of developing acute exacerbations, P. aeruginosa is unlikely to play a role in bacterial-infection-induced fibrosis exacerbation in patients with IPF, and therefore may be a less relevant pathogen to examine bacterial-infection-induced fibrosis exacerbation in mice. Second, different mouse models of lung fibrosis were employed to examine bacterial-infection-induced fibrosis exacerbation, that is, bleomycin versus AdTGFβ1- and SPC-DTR-induced lung fibrosis. Third, we here employed a clinical serotype 19 S. pneumoniae strain known to cause focal pneumonia in mice not progressing towards bacteraemia and sepsis, thereby verifying that all mice reached the endpoint of fibrosis exacerbation analysis at day 7 post-infection, while the recently employed P. aeruginosa infection model also caused bacteraemia in mice, but data on endpoint mortality was not reported. Of note, another important difference between the employed pathogens is that Gram-negative P. aeruginosa does not produce Ply, which is a pneumococcus-specific cell-associated virulence factor.35
In summary, using two independent lung fibrosis models, we show for the first time that Gram-positive bacterial infection with S. pneumoniae causes progression of established lung fibrosis in mice. The pneumococcal virulence factor pneumolysin was identified to be causative for increased fibrogenesis in experimental fibrosis in a TLR4-independent manner, thus supporting its central role in mediating Spn-induced fibrosis exacerbation in mice. Antibiotic treatment as well as vaccination abolished infection-induced fibrosis exacerbation in mice. The current data strongly support both standardised antibiotic treatment and pneumococcal vaccination in patients with lung fibrosis to prevent infection-induced acute exacerbation of lung fibrosis.
Acknowledgments
We thank M Blaess and A-K Janze for excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
SK, BU, MK and UAM are joint first or senior authors of this paper.
Contributors SK and BU performed the experiments, analysed the data and wrote the manuscript. RM, JB, ND, MTT, H-GH, DJ, NI, JCP, ADO, SL and MB performed the experiments. TW, WS, AG, THS, JG, MK and UAM designed the study, analysed the data and wrote the manuscript.
Funding This work was supported by grants from the German Research Foundation (DFG), grant A12, to UAM and TW, and by grants of the German Federal Ministry of Education and Research (BMBF) for support of the German Center for Lung Research (DZL) and the Center for Sepsis Control and Care. MK and JG received funding from the Canadian Institute for Health Research (CIHR).
Competing interests None declared.
Ethics approval All animal experiments were approved by local government authorities.
Provenance and peer review Not commissioned; externally peer reviewed.