Article Text

Abstract
Background Mesenchymal stromal cells (MSCs) demonstrate considerable promise in preclinical acute respiratory distress syndrome models. We wished to determine the efficacy and mechanisms of action of human MSCs (hMSCs) in the setting of acute lung injury induced by prolonged Escherichia coli pneumonia in the rat.
Methods Adult male Sprague Dawley rats underwent intratracheal instillation of E. coli bacteria in all experiments. In Series 1, animals were randomised to intravenous administration of: (1) vehicle (phosphate buffered saline (PBS), 300 μL); (2) 1×107 fibroblasts/kg; (3) 1×107 hMSCs/kg or (4) 2×107 hMSCs/kg. Series 2 determined the lowest effective hMSC dose. Series 3 compared the efficacy of intratracheal versus intravenous hMSC administration, while Series 4 examined the efficacy of cryopreserved hMSC. Series 5 examined the efficacy of the hMSC secretome. Parallel in vitro experiments further assessed the potential for hMSCs to secrete LL-37 and modulate macrophage phagocytosis.
Results hMSC therapy reduced the severity of rodent E. coli pneumonia, improving survival, decreasing lung injury, reducing lung bacterial load and suppressing inflammation. Doses as low as 5×106 hMSCs/kg were effective. Intratracheal hMSC therapy was as effective as intravenous hMSC. Cryopreserved hMSCs were also effective, while the hMSC secretome was less effective in this model. hMSC therapy enhanced macrophage phagocytic capacity and increased lung and systemic concentrations of the antimicrobial peptide LL37.
Conclusions hMSC therapy decreased E. coli induced pneumonia injury and reduced lung bacterial burden, potentially via enhanced macrophage phagocytosis and increased alveolar LL-37 concentrations.
- ARDS
- Bacterial Infection
Statistics from Altmetric.com
Key messages
What is the key question?
Are human derived bone marrow mesenchymal stromal cells (MSCs) capable of decreasing Escherichia coli induced acute lung injury (ALI) and how might these effects be mediated?
What is the bottom line?
Human bone marrow MSCs decreased E. coli induced ALI, in part by enhancing the host antimicrobial response.
Why read on?
This study highlights the therapeutic potential of human MSCs (hMSCs) to decrease E. coli induced ALI. hMSC therapy improved animal survival, reduced lung bacterial counts, decreased physiological and histological evidence of injury, was effective when administered intravenously or intratracheally, and was effective after undergoing cryopreservation and storage. These effects, which were not seen with fibroblasts, may have been mediated in part via enhancement of macrophage function and increased antimicrobial peptide production.
Introduction
Acute respiratory distress syndrome (ARDS) constitutes a spectrum of severe acute respiratory failure characterised by an acute onset of severe hypoxia within 1 week of a precipitating event, changes indicative of widespread airspace disease on chest radiograph or CT, and permeability pulmonary oedema.1 When ARDS occurs in the setting of multisystem organ failure, mortality rates over 60% have been reported and ARDS is the leading cause of death and disability in the critically ill.2 There are no specific therapies for ARDS, and management remains supportive. ARDS develops most commonly in the context of severe sepsis,3 particularly infection with Gram-negative bacilli such as Escherichia coli4 and sepsis-induced ARDS has the worst outcome.5
Mesenchymal stem cells /mesenchymal stromal cells (MSCs) constitute a promising therapeutic strategy for ARDS.6 Human MSCs (hMSCs) express low levels of human leucocyte antigen (HLA) class I and do not express HLA class II, allowing them to bypass T cell immune responses and permit allotransplantation and xenotransplantation.7 hMSCs suppress the proliferation, interferon γ production and cytotoxicity of CD4+ and CD8+ T cells, induce the generation of T regulatory cells, suppress B cell responses including immunoglobulin production and impair antigen presentation by dendritic cells. Our current understanding of MSC-mediated immunomodulation in ARDS and sepsis suggests that hMSCs induce an ‘anti-inflammatory, pro-repair’ macrophage phenotype (M2) that appears central to its mechanism of action.8 ,9 Of interest, hMSCs also appear to enhance macrophage phagocytosis and killing of bacteria, features which are considered more typical of the proinflammatory (M1) phenotype.10 MSCs demonstrate beneficial effects in preclinical lung injury models including bleomycin induced acute lung injury (ALI),11 Bleomycin induced fibrosis,12 ,13 abdominal sepsis,8 ,14 ,15 ventilator induced lung injury16 ,17 and sterile pneumonia.18–20
Recent studies have demonstrated that human derived MSCs (hMSCs) are effective in human lungs ex vivo21 and can reduce mortality in murine14 and ovine22 sepsis models. A phase 1, open label, dose escalation, multicentre clinical trial has recently demonstrated the safety of allogeneic BM-MSCs in patients with moderate to severe ARDS.23 We have demonstrated that hMSCs can enhance repair and recovery of function following ventilator induced lung injury.24 We therefore wished to further characterise the therapeutic potential of hMSCs in a relevant preclinical model of E. coli induced ARDS. We performed a series of studies to test the following set of hypotheses: (1) that hMSCs would reduce E. coli induced lung injury and inflammation; (2) that a dose response would exist for hMSC therapy; (3) that the intravenous and intratracheal routes of administration would prove similarly effective; (4) that hMSCs would retain efficacy following storage and cryopreservation; (5) the hMSC secretome would reduce E. coli induced injury; and (6) that hMSC therapy would exert its beneficial effect by enhancing the host antimicrobial defenses.
Materials and methods
All work was approved by the Animal Care Research Ethics Committee of the National University of Ireland, Galway and conducted under license from the Department of Health, Ireland. Specific-pathogen-free adult male Sprague Dawley rats (Charles River Laboratories, Kent, UK) weighing between 350 g and 450 g were used in all experiments. A full description of the methods is available in the online supplementary digital content 1.
Preparation of cells and conditioned medium
The hMSCs used in these studies were provided by Orbsen Therapeutics (Galway, Ireland). Briefly, bone marrow was harvested from volunteers (see online supplementary table A, digital content 2), filtered, centrifuged, and the cell pellets cultured as previously described.25 Adherent cells were further expanded until 80% confluent, and then trypsinised and culture expanded to passage 4, whereupon they were used for experiments. For one series (see below) hMSCs were cryopreserved in 10% DMSO/foetal bovine serum (FBS) freezing solution in aliquots of 5×106 cells, and later thawed prior to use, and either administered directly (‘Thawed hMSCs’) or first washed to remove cyropreservative solution and administered (‘Washed hMSCs’). Primary human lung fibroblasts and the U937 monocytic/macrophage cell line were purchased from American Type Culture Collection (ATCC, Manassas, Virginia, USA).
For generation of conditioned medium (CM), hMSCs (4×106 cells) were washed, cultured in serum-free medium for 24 h, then rewashed, and fed again. The serum-free CM was subsequently harvested at 24 h (CM-24 h) or 48 h (CM-48 h). For in vivo experiments 15 mL of this medium was concentrated using a 3000 Dalton centrifugal concentrating filter (Amicon, Billerica, Massachusetts, USA) to give a final volume of 300µL.
Rodent E. coli induced injury protocol
E. coli instillation: The E. coli used in these experiments was E5162 (serotype: O9 K30 H10) and was supplied by the National Collection of Type Cultures, Central Public Health Laboratory, London, UK.26 ,27 Preliminary experiments were performed to determine the bacterial load of intratracheal E. coli required to produce a lung injury over a 48 h period. Animals were anaesthetised by inhalational induction with isoflurane and intraperitoneal ketamine 40 mg/kg (Pfizer, Kent, UK). Following confirmation of anaesthesia depth, 2×109 E. coli in a 300 μL phosphate buffered saline (PBS) suspension was instilled into the trachea under direct vision, and the animals allowed to recover.28
Assessment of lung injury: Forty-eight hours after E. coli instillation, animals were reanaesthetised as described above and anaesthesia maintained with Saffan (Alfaxalone 0.9% and alfadolone 0.3%, Schering Plough, Welwyn Garden City, UK). A tracheostomy tube was inserted and intra-arterial access was sited in the carotid artery. Muscle relaxation was induced with cisatracurium besylate, and the lungs were mechanically ventilated with 30% O2 in 70% N2. Systemic arterial blood pressure and peak airway pressure were continually measured. Body temperature was maintained at 36–37.5°C. Lung static compliance and arterial blood gas analysis were measured after 20 min and were repeated on 100% O2 after 15 min.29 ,30 The physiological assessment of lung function (oxygenation, lung compliance) was performed by unblinded investigators. Animals were then killed by exsanguination under anaesthesia.
Ex vivo analyses: Ex vivo analyses were restricted to animals that survived the experimental protocol. Briefly, the heart-lung block was dissected from the thorax, bronchoalveolar lavage (BAL) was performed, and BAL fluid differential leucocyte counts and lung bacterial colony counts completed. BAL fluid was centrifuged, and the supernatant was snap frozen and stored at −80°C. BAL concentrations of interleukin (IL) 6, IL-10 and keratinocyte growth factor (KGF) and were determined using ELISA (R&D Systems, TM Abingdon, UK) and BAL protein concentrations measured (Micro BCA Protein 16 assay kit, Pierce, Rockford, Illinois, USA). BAL and serum concentrations of LL-37 were determined (Cambridge Biosciences, Cambridge, UK). The left lung was isolated and fixed, and the extent of histological lung damage determined using quantitative stereological techniques.31 All ex vivo analyses (BAL analyses, Wet:Dry ratios, histological analyses) were performed by blinded investigators.
Experimental design
Series 1 examined the efficacy of hMSC therapy in attenuating E. coli induced lung injury. Thirty minutes following intratracheal instillation of E. coli bacteria, animals were randomised to intravenous administration of: (1) vehicle (PBS, 300 μL); (2) 1×107 fibroblasts/kg; (3) 1×107 hMSCs/kg or (4) 2×107 hMSCs/kg. Series 2 evaluated the lowest effective hMSC doses, with animals randomised to receive: (1) vehicle (PBS, 300 μL); (2) 1×107 hMSCs/kg; (3) 5×106 hMSCs/kg or (4) 2×106 hMSCs/kg. Series 3 compared the efficacy of intratracheal versus intravenous hMSC administration, with animals randomised to receive: (1) Intravenous vehicle (PBS, 300 μL); (2) Intravenous 1×107 hMSCs/kg; (3) Intratracheal 1×107 hMSCs/kg or (4) Intratracheal vehicle (PBS, 300 μL). Series 4 examined the efficacy of cryopreserved hMSCs, with animals randomised to receive: (1) Intravenous vehicle (PBS, 300 μL); (2) Intravenous 1×107 fresh hMSCs/kg; (3) Intravenous 1×107 thawed cryopreserved hMSCs/kg (‘Thawed hMSCs’); and (4) Intravenous 1×107 cryopreserved hMSCs/kg thawed and washed prior to use (‘Washed hMSCs’). Series 5 examined the efficacy of the hMSCs secretome, with animals randomised to receive: (1) Intravenous vehicle (PBS, 300 μL); (2) Intravenous 1×107 hMSCs/kg; (3) medium conditioned for 24 h with hMSCs (CM-24 h); and (4) medium conditioned for 48 h with hMSCs (CM-48 h). In all experiments, the severity of the lung injury was assessed after 48 h.
Effect of hMSC on monocyte and macrophage phagocytosis
Peripheral blood monocytes were isolated from whole blood, from animals that received hMSC or vehicle therapy, by Ficoll gradient and the effect of hMSC therapy on phagocytic capacity determined. U937 cells, differentiated into macrophages via exposure to phorbol myristate acetate 1 μg/mL for 72 h, were seeded in six-well plates with or without hMSCs and exposed to E. coli bacteria for 4 h. The phagocytic potential of the rodent monocytes and U937-derived macrophages was determined using the Vybrant Phagocytosis Kit (Life Technologies, New York, USA).
Statistical analysis
Data was analysed using Sigma Stat (SYSTAT software, Richmond, California, USA). The distribution of all data was tested for normality using Kolmogorov-Smirnov tests. Animal survival was analysed using the logrank test, with combined hMSC treated groups compared with combined control groups within each series. In series 1, 2 and 4, data were analysed by one-way analysis of variance (ANOVA), with post hoc testing using Dunnett's test, with the vehicle group as the comparison group or by Kruskal-Wallis ANOVA on ranks with post hoc testing using Dunn's method for non-normally distributed data. In series 3, which examined the efficacy of different routes of hMSC administration, a two-way ANOVA was used, with treatment (hMSCs vs vehicle) and route of administration (intravenous vs intratracheal) as the two factors. Subsequent between-group analyses, where indicated, were restricted to comparisons of hMSCs versus vehicle for each route of administration. Underlying model assumptions were deemed appropriate on the basis of suitable residual plots. A two-tailed p value <0.05 was considered statistically significant.
Results
Series 1: Efficacy of hMSC in E. coli induced lung injury
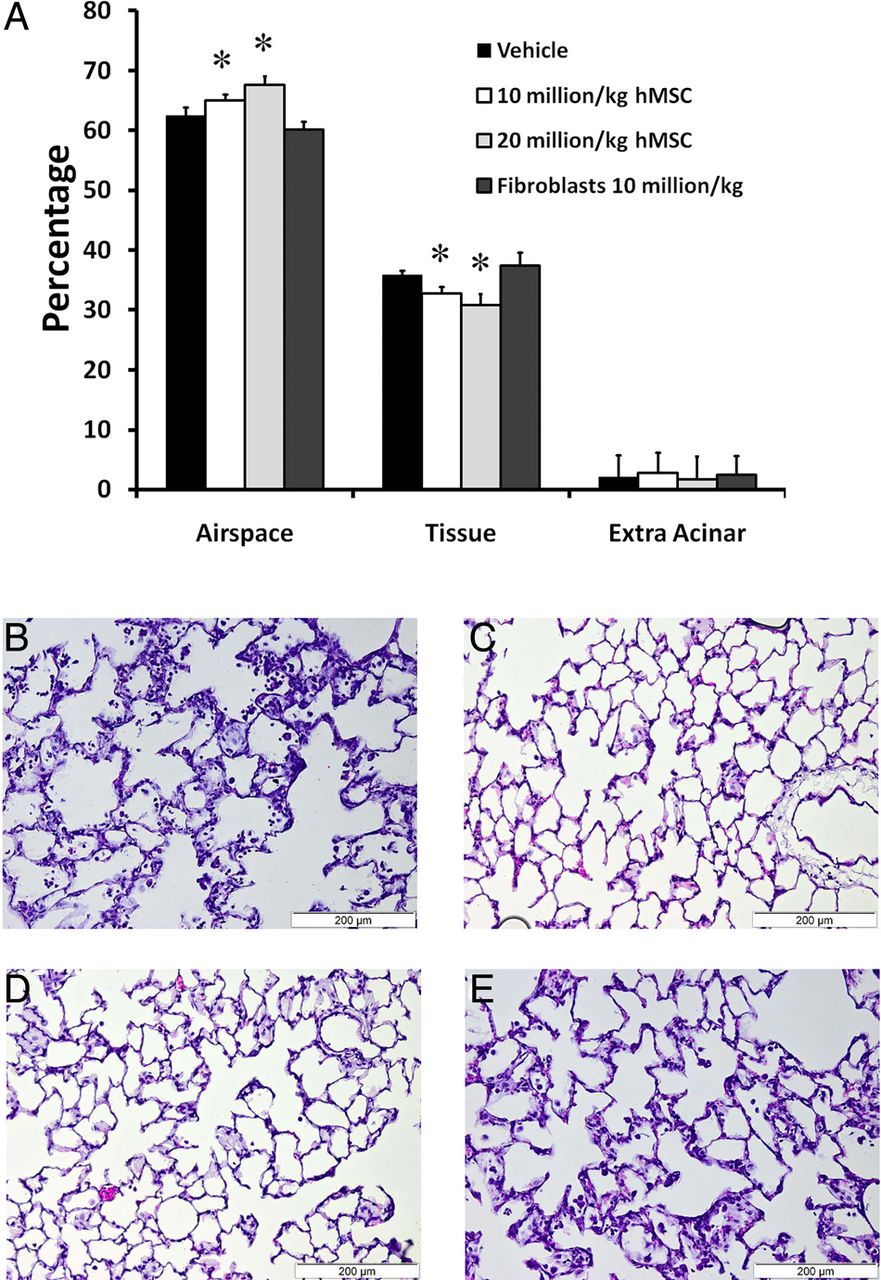
Forty-eight animals were entered into this experimental series, with 12 animals allocated to each group. There were no significant differences between the groups at baseline in terms of preinjury variables, or the amount of instilled E.coli bacteria. One hundred per cent of animals treated with hMSC survived, compared with 71% in the vehicle and fibroblast therapy groups (figure 1A). Both doses of hMSC therapy reduced the severity of physiological lung injury caused by E. coli, increasing arterial oxygenation (figure 1B) and lung compliance (figure 1C). Of interest, only the 20 million hMSCs dose decreased alveolar fluid protein concentrations (figure 1D). While both hMSC doses decreased BAL neutrophils (figure 1E) only the higher hMSC dose reduced E. coli bacterial counts (figure 1F). hMSC therapy had no effect on alveolar IL-6 (figure 1G), but increased alveolar IL-10 (figure 1H) and KGF (figure 1I) concentrations. Ten million hMSCs/kg and 20 million hMSCs/kg reduced histological injury, decreasing alveolar thickening and increasing airspace volume compared with vehicle or fibroblast therapy (figure 2A). Representative histological sections of lung demonstrate the greater degree of resolution of injury and alveolar infiltrates in the hMSC-treated animals (figure 2B–E).

Escherichia coli induced lung injury is decreased by 10–20 million hMSCs/kg. The 10 million hMSCs/kg and 20 million hMSCs/kg doses enhanced animal survival (A), increased mean arterial oxygenation (B) and increased mean static lung compliance (C), 48 h following pulmonary E. coli instillation, compared with either vehicle or fibroblast therapy. Only the higher dose decreased mean BAL protein concentrations (D), both doses decreased neutrophil counts (E), while the higher dose reduced median lung E. coli bacterial load (F). hMSCs did not modulate mean BAL IL-6 (G), while both hMSC doses increased mean BAL IL-10 (H) and mean keratinocyte growth factor (KGF) (I), compared with vehicle or fibroblast therapy. BAL, bronchoalveolar lavage; hMSC, human mesenchymal stromal cell; Vehicle, treatment with vehicle alone. Error bars represent SD. n=12 animals per group. *Significantly (p<0.05) different from the vehicle-treated group.

hMSC therapy decreases the severity of structural lung injury. hMSC therapy enhanced resolution of histological injury as evidenced by decreased mean alveolar lung tissue and increased mean alveolar airspace fractions (A). Representative photomicrographs of lung from a vehicle treated (B), 20 million hMSCs/kg treated (C), 10 million hMSCs/kg treated (D) and a fibroblast treated (E) animal demonstrate greater resolution of lung injury with MSCs at 24 h (n=12 animals per group). Scale bar is 200 μm. hMSC, human mesenchymal stromal cell. Error bars represent SD. *Significantly (p<0.05) different from the vehicle-treated group.
Additional data for this and subsequent experimental series is available in tables B–F in the supplemental digital content 2.
Series 2—Determination of the lowest effective hMSC dose
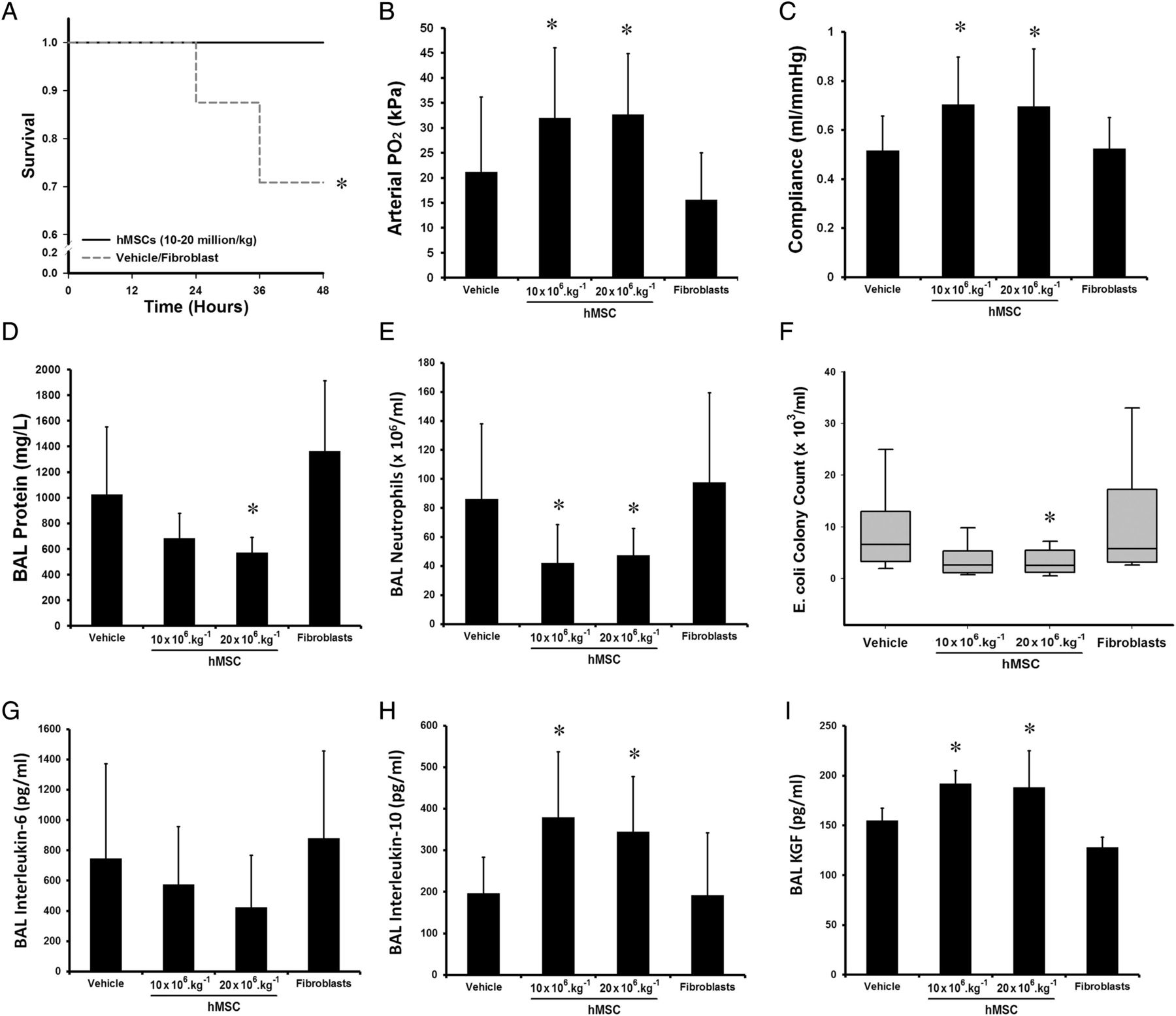
Sixty animals were entered into this experimental series. Treatment with hMSCs enhanced animal survival, with survival rates of 100% in the 5 million/kg (n=14/14) and 10 million/kg (n=14/14) hMSC groups, 93% (n=13/14) in the 2 million/kg group while only 78% (14/18) of vehicle animals survived (figure 3A). The 10 million hMSCs/kg dose was most effective in reducing E. coli induced lung injury. The 5 million hMSCs/kg and 10 million hMSCs/kg doses improved arterial oxygenation (figure 3B), with only the higher dose improving lung compliance (figure 3C), while no dose significantly reduced BAL protein concentrations (figure 3D). Ten million hMSCs/kg decreased alveolar neutrophil counts (figure 3E). All doses of hMSCs decreased E. coli lung bacterial burden (figure 3F) and alveolar IL-6 concentrations (figure 3G), while only the 10 million hMSCs/kg dose increased alveolar IL-10 (figure 3H) and KGF (figure 3I) concentrations and reduced E. coli induced histological injury.

Effect of 2–10 million hMSCs/kg on severity of Escherichia coli induced lung injury. hMSC therapy enhanced animal survival (A). The 10 million hMSCs/kg dose was most effective, enhancing mean arterial oxygenation (B), increasing mean static lung compliance (C), had no effect on mean BAL protein concentrations (D), decreasing mean alveolar neutrophil counts (E), reducing median lung E. coli bacterial load (F), decreasing mean BAL IL-6 (G), and increasing mean BAL IL-10 (H) and KGF (I) compared with vehicle. The 5 million hMSCs/kg and 2 million hMSCs/kg doses were less effective, but did reduce lung E. coli counts, and IL-6 concentrations, while the 5 million hMSCs/kg dose increased arterial PO2 compared with vehicle. BAL, bronchoalveolar lavage; hMSC, human mesenchymal stromal cell; Vehicle, treatment with vehicle alone. Error bars represent SD. n=14–18 animals per group. *Significantly (p<0.05) different from the vehicle-treated group.
Series 3—Determination of optimal route of hMSC delivery
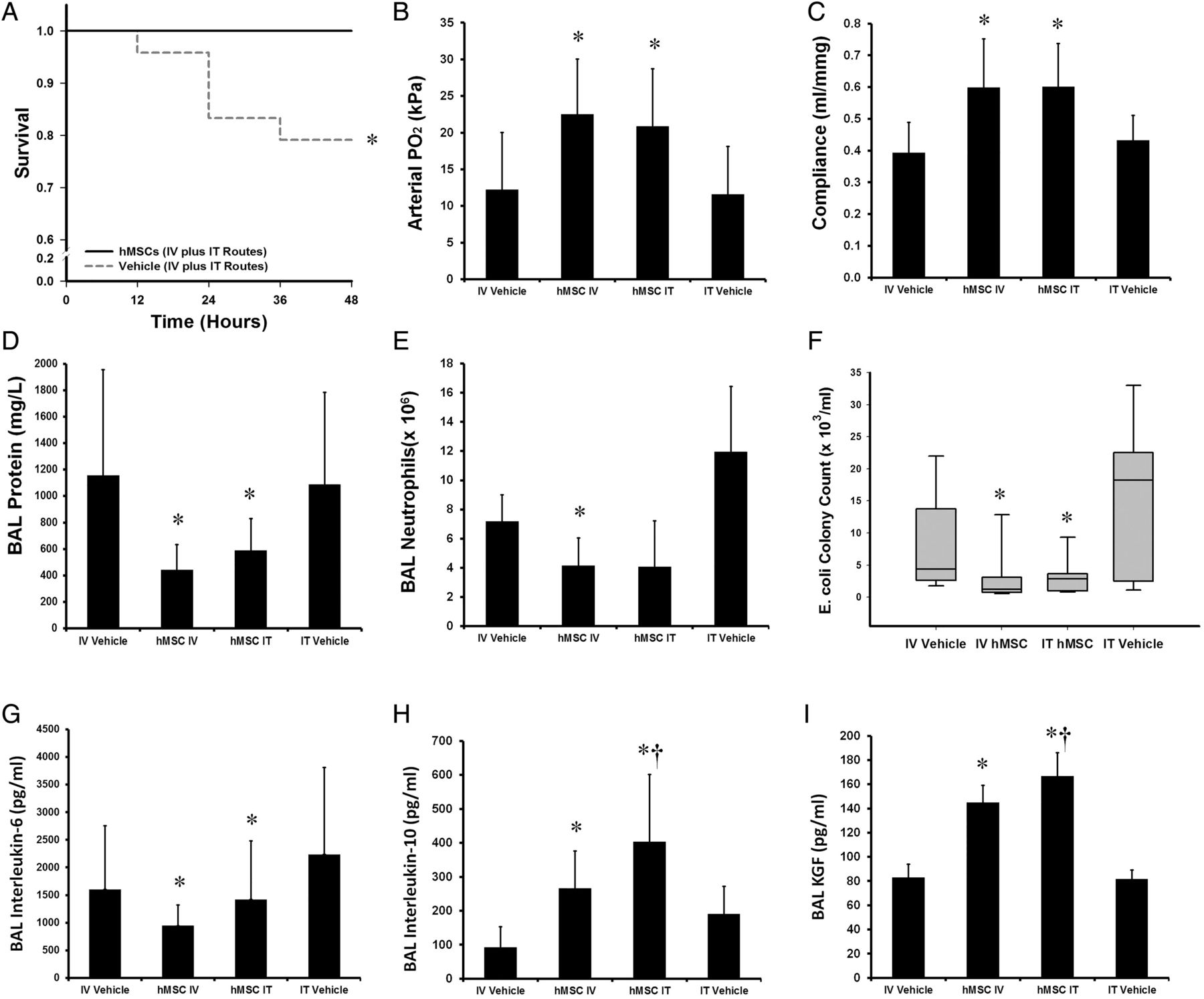
Forty-eight animals were entered into this experimental series, with 12 animals allocated to each group. There were no significant differences between the groups at baseline. Intravenous and intratracheal hMSC therapy significantly enhanced animal survival (figure 3A) and were equally effective in reducing E. coli induced lung injury. Intravenous and intratracheal hMSC therapy increased arterial oxygenation (figure 4B) and lung compliance (figure 4C), and decreased alveolar fluid protein (figure 4D). Intravenous — but not intratracheal — hMSCs decreased alveolar neutrophil counts (figure 4E). Intravenous and intratracheal hMSCs significantly decreased E. coli lung bacterial burden (figure 4F), decreased alveolar IL-6 (figure 4G), and increased alveolar IL-10 (figure 4H) and KGF (figure 4I) concentrations. Of interest, intratracheal hMSC therapy was more effective than intravenous hMSC in increasing alveolar IL-10 and KGF concentrations.

Intratracheal hMSC delivery and intravenous hMSC delivery comparably decreased Escherichia coli induced lung injury. Intravenous and intratracheal hMSC therapy enhanced animal survival (A) IV and intratracheal (IT) hMSC therapy enhanced mean arterial oxygenation (B) and increased mean static lung compliance (C), decreased mean BAL protein concentrations (D). IV—but not IT—hMSCs reduced mean alveolar neutrophil counts (E) while IV and IT hMSCs reduced median lung E. coli bacterial load (F). Both hMSC delivery routes decreased mean BAL IL-6 (G), increased mean BAL IL-10 (H) and KGF (I) compared with vehicle therapy. BAL, bronchoalveolar lavage; hMSC, human mesenchymal stromal cell; IV, intravenous; IT, intratracheal; Vehicle, treatment with vehicle alone. Error bars represent SD. n=12 animals per group. *Significantly (p<0.05) different from the vehicle-treated group. †Different from the MSC IV group.
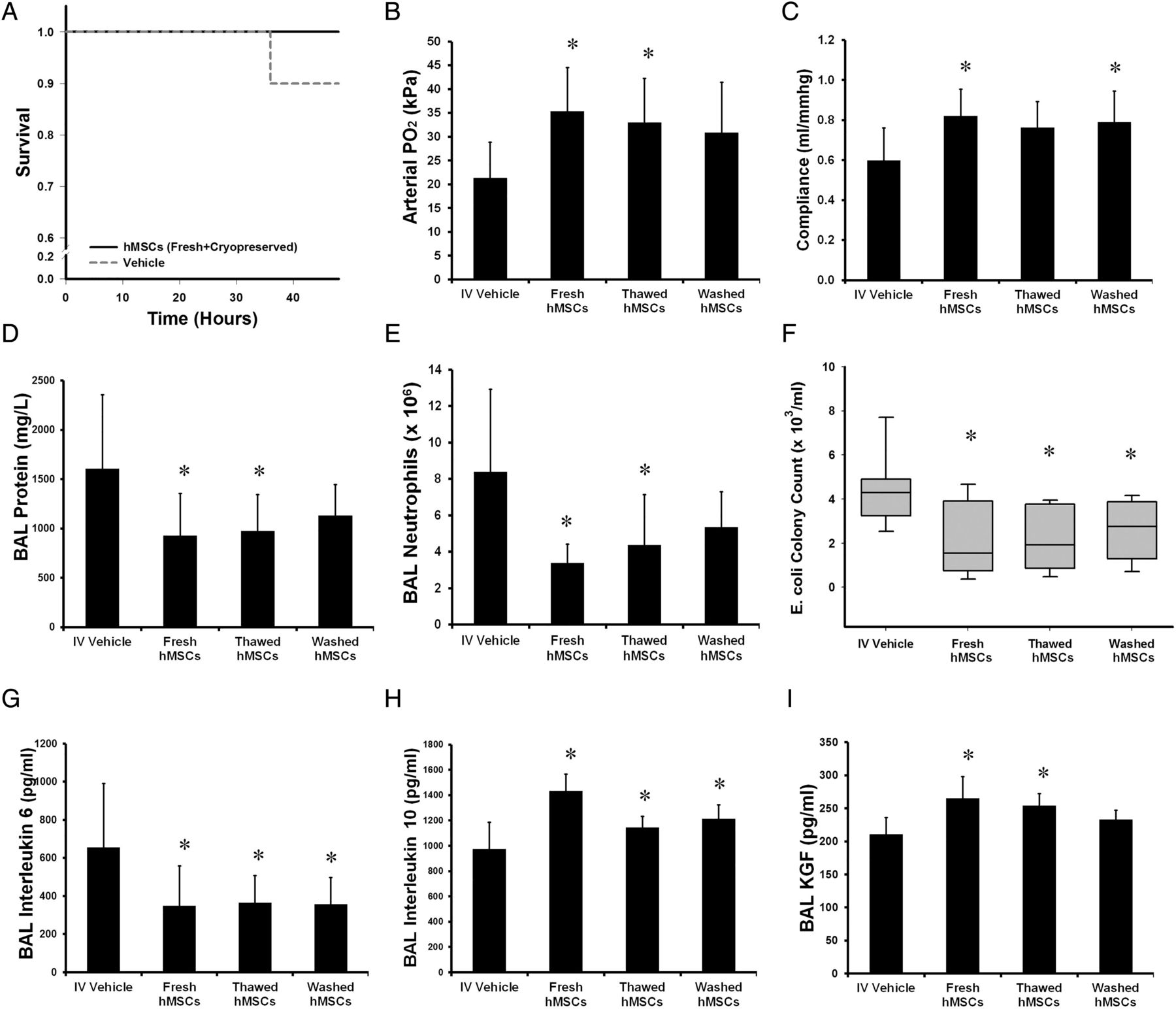
Series 4—Efficacy of cryopreserved hMSC
Forty animals were entered into this experimental series, with 10 animals allocated to each group. There were no significant differences between the groups at baseline. One vehicle-treated animal did not survive. Cryopreservation modestly decreased cell viability from 95.1%±0.6% (fresh hMSCs) to 91.8%±0.6% (cryopreserved hMSCs). Cryopreserved hMSCs, whether administered directly following thawing (‘Thawed hMSCs’), or washed after thawing to remove preservatives (‘Washed hMSCs’) were similarly effective in attenuating E. coli induced injury. hMSC therapy did not alter mortality (figure 5A). Fresh and thawed hMSCs increased arterial oxygenation (figure 5B), while fresh and washed hMSCs improved lung compliance (figure 5C). Fresh and thawed hMSCs decreased alveolar fluid protein (figure 5D), and reduced alveolar neutrophils (figure 5E). Fresh and cryopreserved hMSCs decreased E. coli lung bacterial burden (figure 5F). Fresh and cryopreserved hMSCs decreased alveolar IL-6 (figure 5G), increased alveolar IL-10 (figure 5H), while the fresh and thawed hMSC groups increased alveolar and KGF (figure 5I) concentrations.

Cryopreserved hMSCs decrease Escherichia coli induced lung injury. hMSC therapy, whether administered fresh from culture or following thawing of cryopreserved cells, did not alter survival (A), but did reduce injury as evidenced by enhanced mean arterial oxygenation (B) and increased mean static lung compliance (C), decreased mean BAL protein concentrations (D), reduced mean alveolar neutrophil counts (E), reduced median lung E. coli bacterial load (F), and reduced mean BAL IL-6 (G), increased mean BAL IL-10 (H) and KGF (I) compared with vehicle. Washing hMSCs following thawing, in order to remove cryopreservation agents, did not enhance their efficacy. BAL, bronchoalveolar lavage; hMSC, human mesenchymal stromal cell; Vehicle, treatment with vehicle alone. Error bars represent SD. n=10 animals per group. *Significantly (p<0.05) different from the vehicle-treated group.
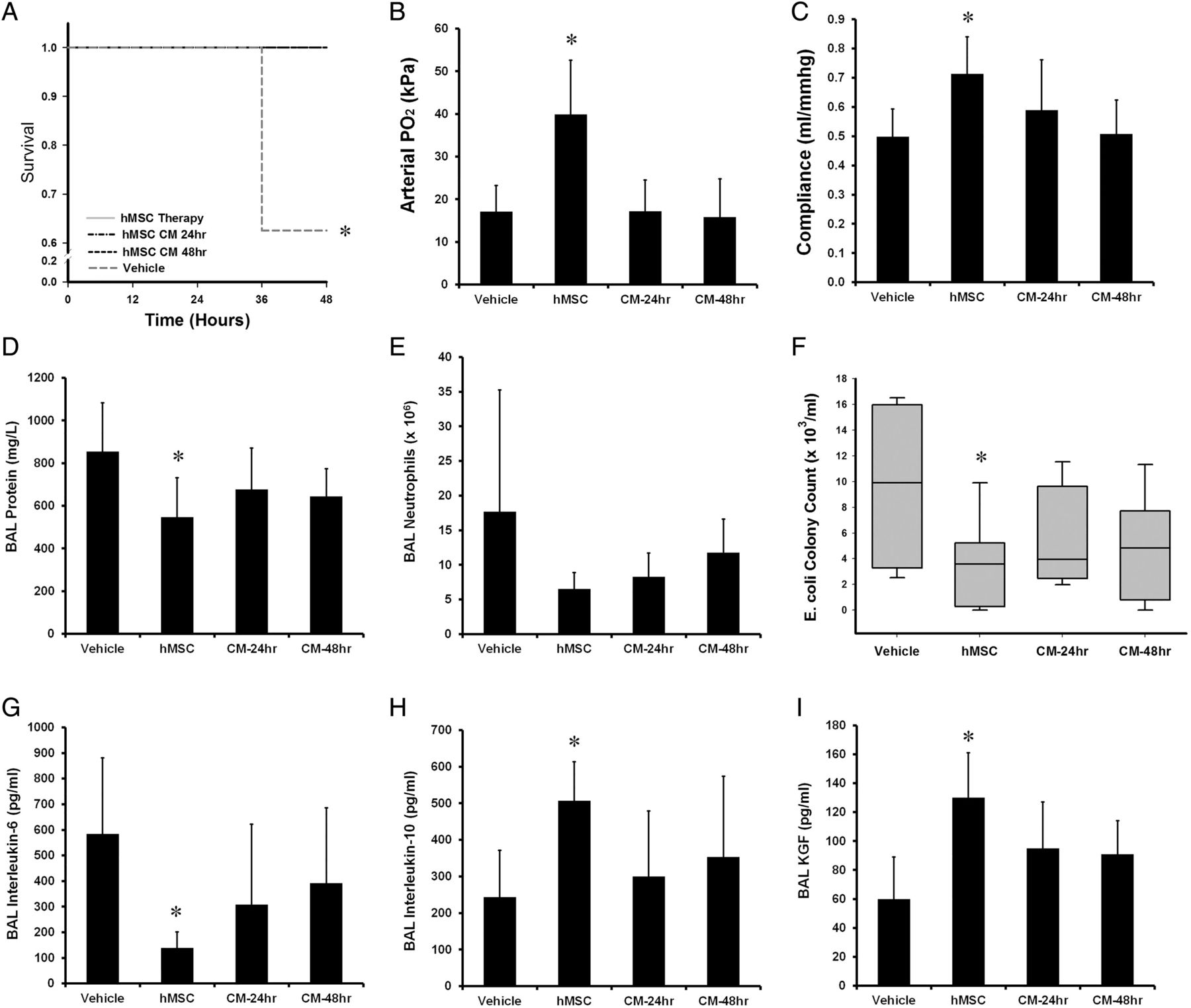
Series 5—Efficacy of the hMSC secretome
Thirty-two animals were entered into this experimental series, with eight animals allocated to each group. There were no significant differences between the groups at baseline. Both hMSC therapy itself, and its secretome (hMSC-CM) enhanced animal survival compared with vehicle (figure 6A). In contrast, neither 24 h nor 48 h hMSC-CM reduced lung injury in surviving animals. hMSC-CM did not increase arterial oxygenation (figure 6B), nor lung compliance (figure 6C), nor reduce BAL protein (figure 6D), BAL neutrophils (figure 6E), nor reduce alveolar E. coli counts (figure 6F). Similarly there was no effect of the secretome on alveolar IL-6 (figure 6G), IL-10 (figure 6E) or KGF concentrations (figure 6F).
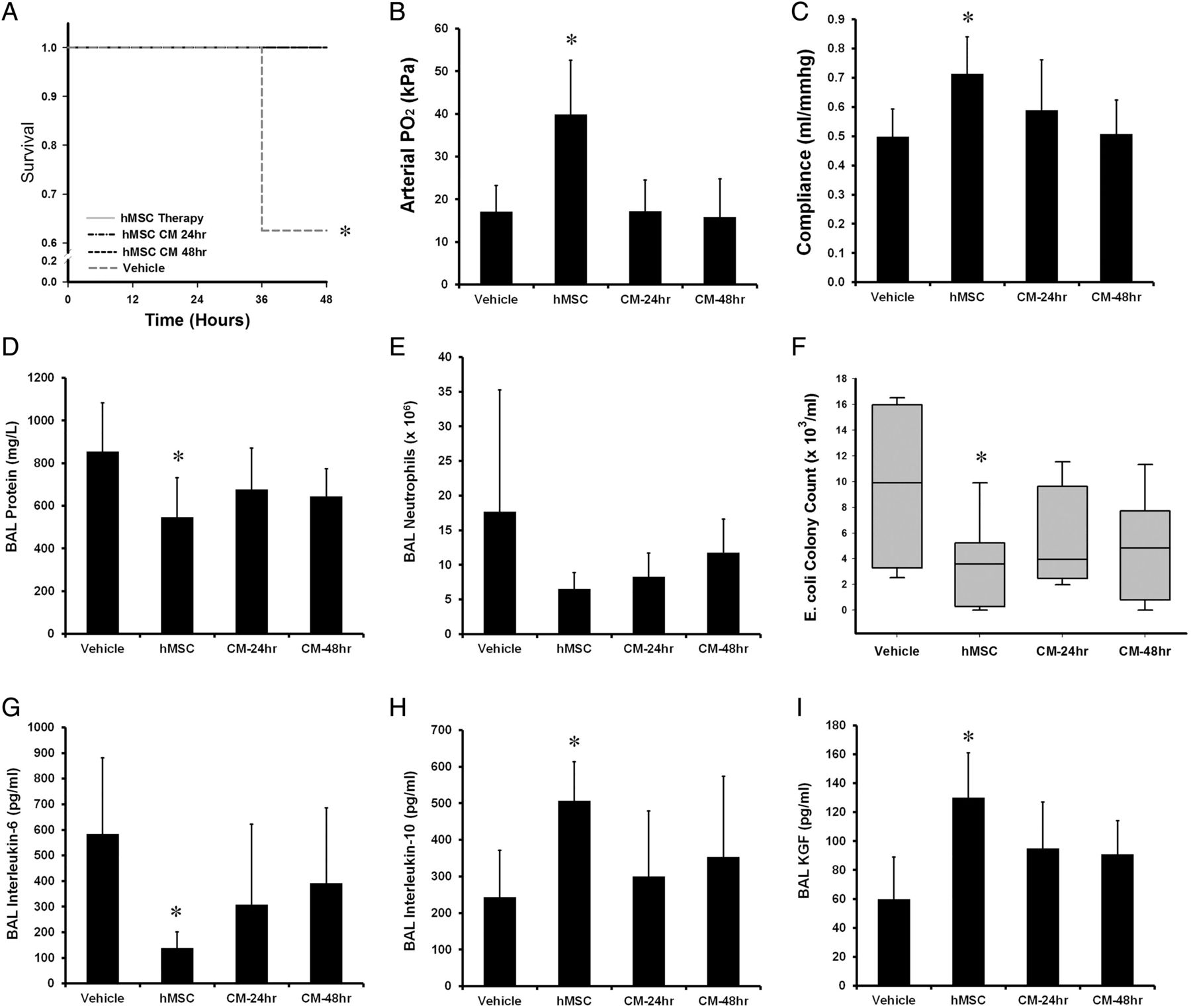
Effects of the hMSC secretome. hMSC therapy and the hMSC conditioned medium (CM) enhanced survival compared with vehicle (A). In contrast, the hMSC-CM did not reduced Escherichia coli injury severity, with no effect on mean arterial oxygenation (B) mean static lung compliance (C), mean BAL protein concentrations (D), mean alveolar neutrophil counts (E), median lung E. coli bacterial load (F) and mean BAL IL-6 (G), IL-10 (H) and KGF (I) compared with vehicle. BAL, bronchoalveolar lavage; CM, conditioned medium; hMSC, human mesenchymal stromal cell; Vehicle, treatment with vehicle alone. Error bars represent SD. n=8 animals per group. *Significantly (p<0.05) different from the vehicle-treated group.
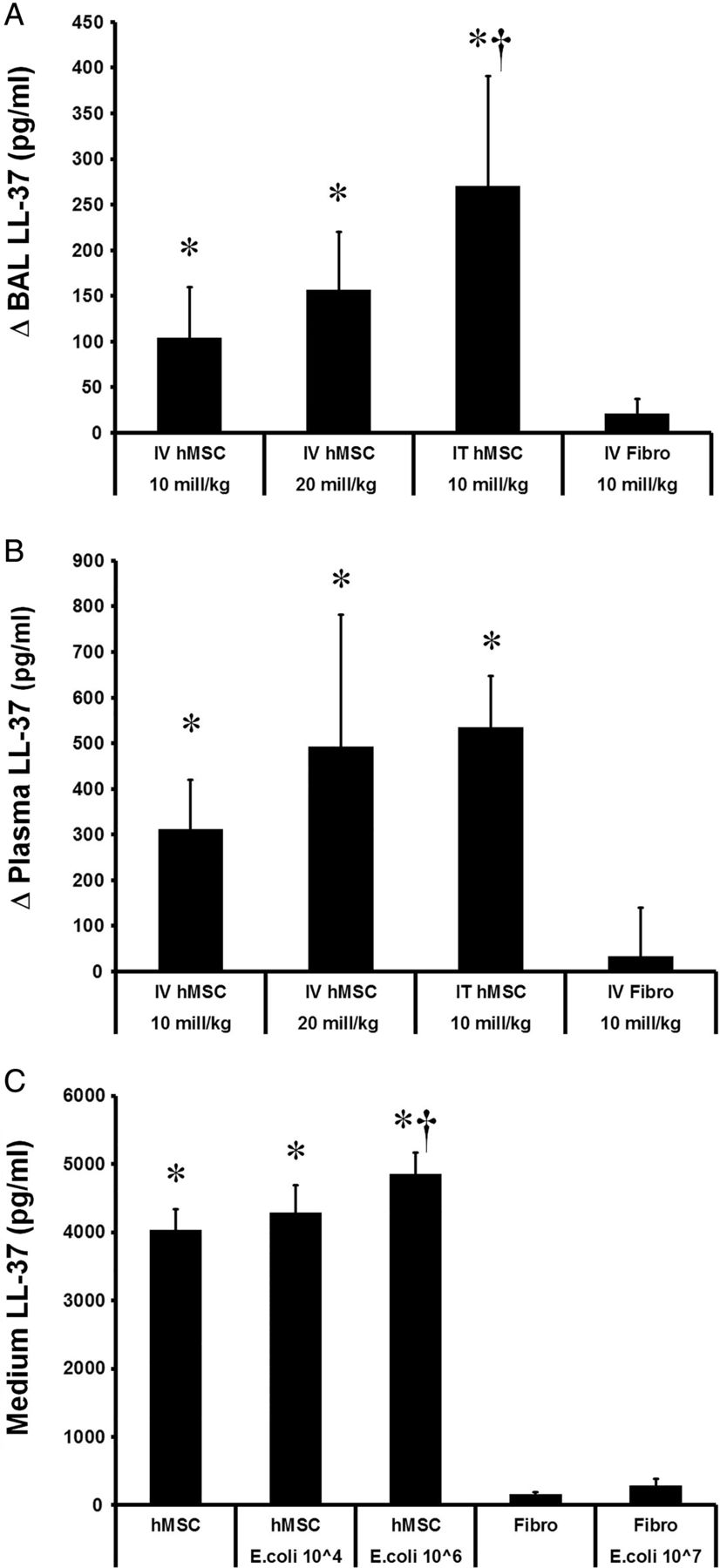
Effect of hMSC therapy on host response to E. coli
hMSC therapy increased alveolar concentrations of the antimicrobial peptide LL-37, whether administered via intravenous or intratracheal routes, compared with fibroblast therapy (figure 7). Delivery of hMSCs via the intratracheal route resulted in higher alveolar concentrations of LL-37 compared with delivery of the same dose intravenously (figure 7A). hMSCs also increased plasma LL-37 concentrations compared with fibroblast treatment, although there was no differential effect of route of delivery (figure 7B). Exposure of hMSCs to E. coli in vitro enhanced LL-37 production, an effect not seen with fibroblasts (figure 7C).

hMSCs produce the antimicrobial peptide LL-37. hMSC therapy increased mean alveolar concentrations of the antimicrobial peptide LL-37 compared with fibroblast therapy (A). Intratracheal (IT) delivery of hMSCs resulted in higher mean alveolar LL-37 concentrations compared with intravenous (IV) delivery (A). Intravenous hMSC therapy and intratracheal hMSC therapy increased mean plasma LL-37 concentrations to a comparable extent (B). Exposure of hMSCs to Escherichia coli in vitro resulted in a dose-dependent increase in mean LL-37 production, an effect not seen with fibroblasts (C). *Significantly (p<0.05) different from the fibroblast-treated group. †Different from the intravenous-hMSC group (Panel A) or different from the hMSC group (Panel C). hMSC, human mesenchymal stromal cell.
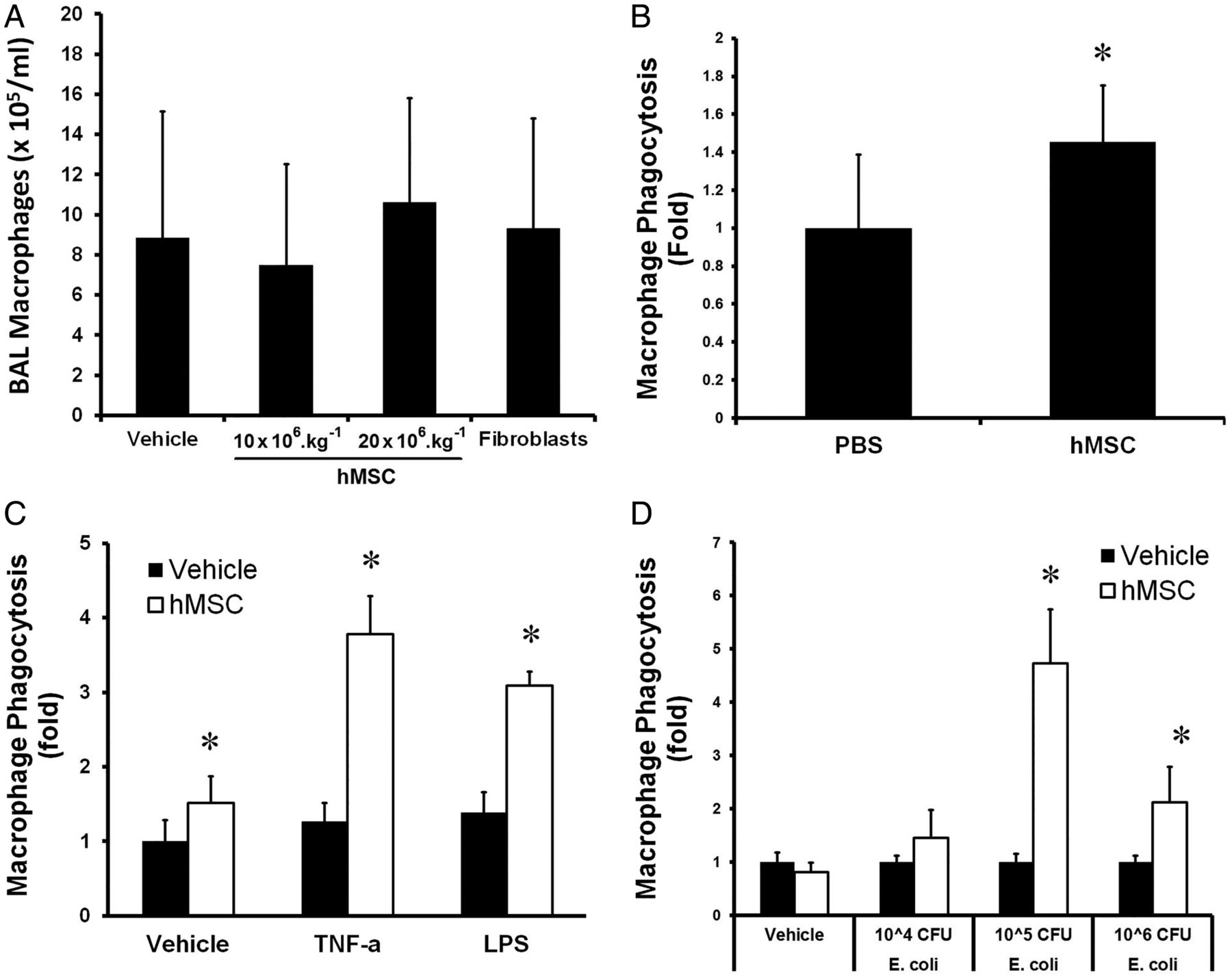
hMSC therapy did not increase the absolute number of macrophages in the lung of E. coli infected animals (figure 8A). Monocytes isolated from the blood of hMSC treated E. coli infected animals demonstrated enhanced bacterial phagocytosis (figure 8B). This effect of hMSCs therapy on phagocytosis was further enhanced when the monocytes were exposed to endotoxin and tumour necrosis factor α (figure 8C). In subsequent in vitro studies, hMSCs enhances the phagocytic capacity of U937 cell derived macrophages exposed to increasing doses of E. coli (figure 8D). Taken together, these studies suggest that hMSCs enhance monocyte/macrophage phagocytosis of bacteria.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
hMSC therapy enhances monocyte/macrophage function. hMSC therapy did not increase the mean number of macrophages in the lung of Escherichia coli infected animals (A). Monocytes isolated from the blood of E. coli infected animals that received hMSCs demonstrated enhanced bacterial phagocytosis (B). This effect was further enhanced when these monocytes were exposed to endotoxin and TNFα (C). hMSCs enhanced the phagocytic capacity of U937 cell derived macrophages exposed to increasing doses of E. coli in vitro (D). BAL, bronchoalveolar lavage; CFU, colony forming unit; hMSC, human mesenchymal stromal cell; LPS, lipopolysaccharide; TNFα, tumour necrosis factor α. *Significantly (p<0.05) different from the vehicle-treated group.
Discussion
In these studies, we demonstrate that hMSCs, transplanted xenogeneically into the immune competent rat reduced lung bacterial counts, and decreased physiological and histological evidence of E. coli induced lung injury. hMSC therapy was comparably effective when administered intravenously or intratracheally. We show for the first time, to our knowledge, that therapeutic efficacy was fully maintained in hMSC following cryopreservation and storage. Of interest, the hMSC secretome, while increasing animal survival, did not reduce E. coli injury. These effects, which were not seen with human lung fibroblast controls, may have been mediated in part via enhancement of macrophage function and increased antimicrobial peptide production. Taken together with our recent finding that hMSCs can enhance repair and recovery of function following ventilator-induced lung injury,24 these data provide important insights regarding the therapeutic potential of human mesenchymal stem cells for ARDS.
hMSCs decreased E. coli induced ALI: hMSC therapy enhanced animal survival and attenuated the severity of E. coli induced ALI. hMSCs decreased physiological indices of lung dysfunction, reducing alveolar-arterial oxygen gradient, increasing lung compliance and decreasing lung permeability, as evidenced by decreased alveolar fluid protein concentrations. hMSC therapy also reduced structural lung injury following E. coli instillation. Fibroblasts did not have any therapeutic effect, suggesting that these beneficial effects are specific to MSCs. These data extend prior findings demonstrating the therapeutic potential of xenogeneic hMSC therapy in murine models of endotoxin32 and bacterial lung injury33 and in the ex vivo human lung.21 We used a rodent E.coli model of lung injury33 ,34 in order to examine the efficacy of xenogeneic hMSC transplantation in immune competent animals, and to determine the effect on lung injury severity in addition to survival and indices of inflammation. These data provide further evidence that the beneficial effects of hMSC are not major histocompatibility complex restricted, underlining the therapeutic potential of allogeneic hMSC in humans. More generally, these results provide further support for the therapeutic potential of hMSC therapy for sepsis.
hMSC dose response: The importance of understanding dose-response characteristics is clear from a recent human COPD trial, which found that a relatively low hMSC dose (1.5 million/kg) was ineffective.35 We found that single hMSC doses as low as 2×106 hMSCs/kg decreased lung bacterial load and resulted in greater animal survival compared with vehicle therapy. Overall, the best balance between efficacy and dose was seen at 1×107 hMSCs/kg. The dose of 5×106 hMSCs/kg did modestly reduce physiological injury, while the 2×106 hMSCs/kg dose did not reduce injury severity. The highest dose studied, 2×107 hMSCs/kg, did not demonstrate clear advantages over the 1×107 hMSCs/kg dose. Consequently we used a dose of 1×107 hMSCs/kg in our subsequent studies. The effective hMSC dose in our studies was similar to that used in prior studies in endotoxin induced injury18 and was considerably less than the hMSC dose used in murine bacterial sepsis studies.33 ,34 While it is difficult to extrapolate from rodent studies to the dose required in humans, doses of 10 million hMSCs/kg are feasible to generate and to administer to patients with ARDS. In fact, this constitutes the upper dose that was successfully administered in the recently reported phase 1, open label, dose escalation, multicentre clinical trial of allogeneic BM-MSCs in patients with moderate to severe ARDS.23
Route of hMSC administration: hMSC therapy was comparably effective when administered intravenously or intratracheally, which extends our earlier findings with rodent MSC therapy16 ,17 and hMSC therapy24 in the setting of repair following VILI. Of interest, while intratracheal administration did increase alveolar IL-10, KGF and LL-37 concentrations, this did not result in reduced bacterial load, decreased lung injury or greater survival. Given the potential risks of intratracheal hMSC administration directly into severely injured lungs, and the absence of clear evidence for an enhanced effect with this route of administration, these findings suggest that the intravenous route may be the preferred option.
Cryopreserved hMSCs: The use of hMSCs in the clinical setting will be greatly facilitated if they can be cryopreserved and stored prior to use, and administered as a thawed cryopreserved product. We found that hMSCs maintained efficacy after undergoing cryopreservation and storage, decreasing E. coli counts, and reducing lung injury severity. Importantly, there was no loss of efficacy compared with the use of fresh hMSCs that had not undergone cryopreservation. Of interest, washing the hMSCs prior to administration, in order to remove cryopreservation agents, did not confer any advantage over the administration of hMSCs directly post thawing. These are important findings from a translational perspective, as they demonstrate that hMSCs can be cryopreserved for transport to clinical sites prior to use, and thawed prior to administration, without loss of efficacy. If confirmed, this would greatly simplify clinical translational studies and eventual use of hMSCs clinically should they prove effective in clinical trials.
hMSC secretome: We found that the hMSC secretome improved animal survival, but did not modulate the severity of E. coli induced lung injury in surviving animals. We have previously demonstrated that the secretome of rodent MSCs enhances repair following VILI, and that this reparative effect was partly KGF mediated.16 Most recently, we have demonstrated that the hMSC secretome enhances repair following established VILI.24 Lee et al have demonstrated that the secretome from hMSCs can attenuate E. coli induced injury to the human lung.21 They further demonstrated that human monocytes expressed the KGF receptor, and that KGF decreased monocyte apoptosis thereby increasing bacterial clearance.
While the beneficial effect of the hMSC secretome on survival is encouraging, the mechanism underlying this beneficial effect is unclear. The hMSC secretome did not reduce bacterial burden and did not modulate alveolar KGF concentrations. The lack of effect of the secretome on alveolar KGF concentrations may explain the lack of effect on bacterial burden, given the importance of KGF in enhancing macrophage phagocytosis.21 ,36 The hMSCs dose we used to generate the secretome corresponds to the 10 million hMSCs/kg dose. Using a higher concentration of hMSCs to generate the hMSC-CM, or administering repeated doses, might have been more effective. Alternatively, non-surviving vehicle-treated animals were likely the worst injured, possibly biasing against detecting a physiological effect of the secretome.
Mechanisms of action of hMSCs: The immunomodulatory effects of hMSC therapy appear important in mediating their effects in reducing E. coli induced lung injury. hMSC therapy reduced alveolar neutrophil infiltration, while increasing alveolar IL-10 concentrations, consistent with previous work using rodent MSCs.9 ,16 Of importance, hMSC therapy reduced lung E. coli bacterial burden, again consistent with previous reports.33 ,34 In keeping with prior studies from our group and others,16 ,25 we found that hMSC therapy increased alveolar concentrations of the growth factor KGF, which has been demonstrated to be important in mediating the effects of hMSC in bacterial injury.21
We focused on two aspects of this antimicrobial response, namely the potential for hMSCs to secrete antimicrobial peptides, and to modulate the monocyte/macrophage response to infection. We found that hMSCs secrete the antimicrobial peptide LL-37, and this antimicrobial peptide is enhanced by exposure to E. coli, while intratracheal hMSC therapy led to higher alveolar—but not plasma—concentrations of LL-37. These findings are consistent with prior reports suggesting that antimicrobial peptides may be a key component of the hMSC response to microbial infection.14 ,34
We found that hMSC therapy did not increase the absolute number of macrophages in the lungs of E. coli infected animals. Monocytes isolated from the blood of hMSC treated E. coli infected animals demonstrated enhanced bacterial macrophage phagocytosis, and these macrophages demonstrated further enhancement of phagocytosis when exposed to endotoxin and tumour necrosis factor α. In subsequent in vitro studies, hMSCs enhanced the phagocytic capacity of macrophages exposed to E. coli. Taken together with our finding that hMSC therapy increases alveolar KGF concentrations, these studies support previous findings suggesting a role for KGF in mediating the MSC-induced monocyte/macrophage phagocytosis of bacteria.21 ,36
Our finding that the presence of E. coli enhanced hMSC LL-37 secretion and also enhanced the effect of hMSC on macrophage function suggests that the hMSC response may be modulated by changes in their microenvironment. This may explain the finding that the hMSC—but not their secretome alone—was effective in reducing E. coli induced injury.
Limitations: There are a number of limitations to these studies. First, our studies were carried out in a rodent model and caution must be exercised in extrapolating to the clinical situation. These studies provide important proofs of concept with regards to hMSC efficacy, dose-response and dose-route characteristics, and efficacy of cryopreserved hMSC rather than providing direct information on how to perform hMSC therapy in humans. Second, we did not provide data on the effect of hMSCs on animals exposed to sham infection. However, we have previously reported that hMSCs do not produce detectable effects on protectively ventilated animals.24 Third, we used macrophages derived from a human monocyte cell line in our in vitro experiments, rather than primary human macrophages. Finally, we have not examined the fate of injected cells, or performed detailed mechanism of action studies. We do provide mechanistic insights regarding the potential for hMSCs to secrete antimicrobial peptides and modulate monocyte/macrophage function. Previously, we found that systemically or intratracheally injected cells accumulate in the lung in the first 24 h, and thereafter are distributed to other organs or the reticuloendothelial system.17
Conclusions
Xenogeneic transplantation of hMSCs decreased E. coli induced pneumonia injury, improved animal survival and reduced lung bacterial burden, potentially via enhanced macrophage phagocytosis and increased alveolar LL-37 concentrations. These studies demonstrate the presence of a non-linear hMSC dose response, show that the intravenous hMSC administration route is as effective as intratracheal administrations, that cryopreserved hMSCs retain efficacy and that the hMSC secretome was less effective in this model. When taken together with other studies examining the effects of hMSCs in relevant preclinical ARDS models, these findings strongly suggest that MSCs may have therapeutic potential for ARDS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
Footnotes
Presentations: Data from this manuscript was presented at the American Thoracic Society Annual Scientific Meetings in May 2013 and 2014.
Contributors JGL, JD, GFC, FB, DO and TO designed the research. JD, DO, SH, SE and CM performed the experiments. JGL and JD analysed the data, drafted the manuscript and are guarantors of the paper.
Funding This work was supported by funding from the European Research Council, Brussels, Belgium, under the Framework 7 Programme (Grant No: ERC-2007-StG 207777) and the Health Research Board, Dublin, Ireland (Grant No: HRB_POR-2011-1061). JGL and GFC are funded by the Canadian Institute of Health Research and the Canadian Anesthesiology Society and hold a Merit Award and a Clinician Scientist Transition Award, respectively, from the University of Toronto Department of Anesthesia.
Competing interests SE is a senior research scientist at Orbsen Therapeutics, Galway, Ireland, a company which is developing mesenchymal stromal cells for therapeutic purposes. TO and FB are founders, directors and equity holders in Orbsen Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement We are happy to share data from this study.