Article Text

Abstract
The rise in life expectancy worldwide has been accompanied by an increased incidence of age-related diseases, representing an enormous burden on healthcare services and society. All vital organs lose function with age, and this is well described in the lung, with a progressive decline in pulmonary function after the age of about 25 years. The lung ages, like any other organ, with progressive functional impairment and reduced capacity to respond to environmental stresses and injury. Normal physiological ageing results in enlarged alveolar spaces and loss of lung elasticity in the elderly known as ‘senile emphysema’, whereas in COPD there is destruction of the alveolar walls and fibrosis of peripheral airways. However, COPD shows striking age-associated features, such as an increase in cellular senescence, stem cell exhaustion, increased oxidative stress, alteration in the extracellular matrix and a reduction in endogenous antiageing molecules and protective pathways such as autophagy. In this review we discuss the evidence showing how oxidative stress induces accelerated ageing by upregulating the phosphatidylinositol-4,5-bisphosphate 3-kinase/AKT/mechanistic target of rapamycin signalling pathway resulting in depletion of stem cells, defective autophagy, reduced antioxidant responses and defective mitochondrial function thus generating further oxidative stress. Understanding the mechanisms of accelerated ageing in COPD may identify novel therapeutic approaches.
- COPD Pharmacology
- Cough/Mechanisms/Pharmacology
Statistics from Altmetric.com
Introduction
Chronic degenerative diseases are a major and increasing medical and societal burden in developed and developing countries yet there are few therapeutic successful approaches to prevent and treat these common conditions. Many of these non-communicable chronic diseases, including COPD, as well as ischaemic heart disease, diabetes and Alzheimer's disease, involve an acceleration of the normal ageing process. There have recently been major advances in understanding the molecular and cellular mechanisms involved in ageing which may lead to the identification of novel therapeutic targets for the development of drugs to halt or even reverse the acceleration of the ageing process.
Current concepts of ageing
Ageing has been defined as “the progressive decline of homoeostasis that occurs after the reproductive phase of life is complete resulting in increased risk of disease or death”.1 Among the evolving theories on ageing, the free radical theory proposes that oxidative stress is a key driver of accelerated ageing and that reactive oxygen species (ROS) formed during normal oxygen metabolism induces damage, the accumulation of which accounts for progressive deleterious changes described as cellular senescence.2 Animal models question this theory as antioxidants fail to prolong life span plus in some species increased ROS levels are important for the normal ageing process.3 Hekimi et al proposed that there is a gradual ROS response which could explain the role of ROS in age-related diseases. Excessive ROS will induce accumulation of molecular damage which shortens life span,4 but an optimal level of ROS is required for healthy ageing.5 The presence of ROS associated with ageing could be the result of the enhanced survival properties that it offers at a younger age.5 However, excessive ROS, greater than that associated with normal ageing, can also induce age-related degeneration favouring chronic inflammation.5 We believe that inhibition of excessive ROS, which causes accelerated ageing, might be beneficial in chronic inflammatory diseases such as COPD.
Cells have a limited number of divisions before they become arrested. The ability that cells have to divide is determined by the exhaustion of programmed proliferative capacity (intrinsic senescence) and by exposure to external factors, such as oxidative stress or ultraviolet light in the case of skin (figure 1A).5 Once damage to DNA is too great to be repaired, cells lose the potential to replicate resulting in cell cycle arrest. Various pathways can precipitate cells into senescence and exhaustion of stem cells (figure 2).5 Williams’ antagonistic pleiotropic theory of ageing proposes that in early life, senescence is a protective mechanism against cancer, but that after procreation it becomes potentially dangerous (figure 1).6 The accumulation of proinflammatory tissue damage combined with an increasingly dysregulated immune system and senescent cells that secrete proinflammatory cytokines is termed ‘inflammaging’.7 Senescent cells remain metabolically active, thus altering their environment for as long as they persist and display what is known as a ‘senescence-associated secretory phenotype’ (SASP) (figure 1).6 ,7 Senescent cells secrete various damaging biochemical and inflammatory mediators thus inducing changes in the surrounding tissue that will eventually negatively impact on the whole organism.6 The accumulation of senescent cells with age resulting in SASP-induced proinflammatory state is known to play a central role in various age-related conditions and is strikingly similar to what is observed in COPD.8

Senescence and loss of stem cell renewal in COPD. (A) Exposure to environmental or exogenous oxidants (represented by ROS) in young or healthy individuals results in cellular damage. Cells respond to mild damage by activating repair mechanisms such as antioxidant, DNA-repair and autophagy. If the damage is too great, cells will undergo senescence thus preventing oncogenic changes. Damaged cells are replaced by stem cells that are activated by senescence signalling. Stem cell renewal plays an important role in tissue regeneration and healing. (B) In older individuals or in those with COPD, excessive ROS will result in increased damage to cells aggravated by a defective repair mechanism. Increased senescent cells and the resulting SASP can induce a paracrine loop feedback system further stimulating inflammation, alveolar destruction, endothelial dysfunction and immunosenescence also increasing the risk of oncogenic changes. Ageing and ROS can also result in loss of stem cells renewal by induced loss of quiescence and stem cell senescence. This would lead to a loss in tissue regeneration. mTOR, mammalian target for rapamycin; ROS, reactive oxygen species; SASP, senescence associated secretory phenotype.
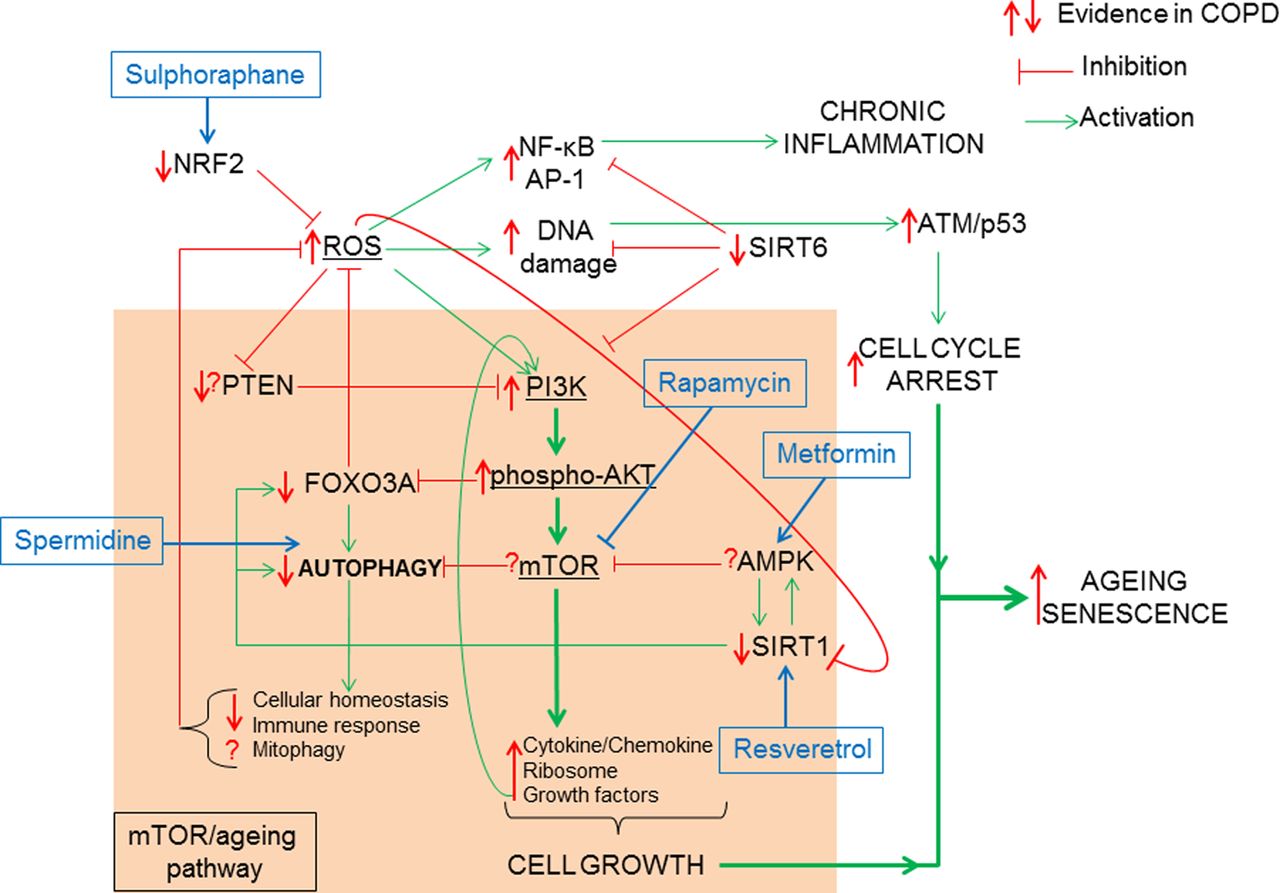
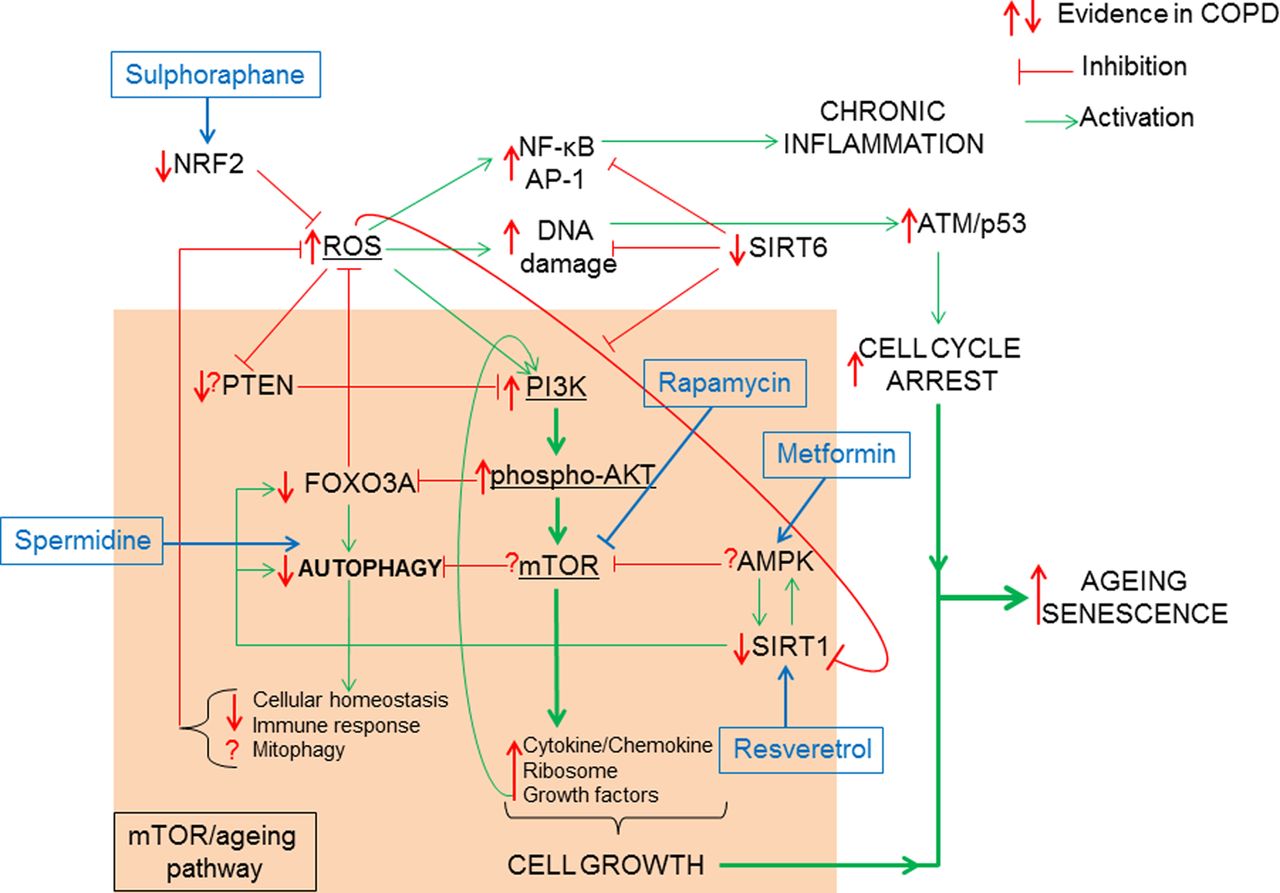
How oxidative stress accelerates the mTOR/ageing pathway. The classic ageing pathway involves the activation of PI3K/phospho-AKT/mTOR by growth factor signalling such as (insulin/insulin growth factor-1 (IGF-1)) resulting in the inhibition of FOXO3A/autophagy and the induction of cytokine and chemokine, ribosomal synthesis and secretion of growth factors favouring cell growth and proliferation. In order to prevent hyperactivation of the mTOR signalling pathway, antiageing molecules such as PTEN inhibit PI3K, AMPK inhibits mTOR and SIRT1 upregulates FOXO3A, thus promoting autophagy. COPD is characterised by elevated ROS possibly linked to a reduction in Nrf2. ROS can activate the mTOR-ageing pathway via PI3K activation. This activation is chronic in COPD due to decreased PTEN and SIRT1 thus resulting in reduced antioxidant defence by FOXO3A inhibition and a loss of autophagy. Loss of autophagy can prevent the clearance of defective mitochondria (mitophagy) further increasing ROS production. In COPD defective autophagy decreases immune response to bacteria and decreases cellular homoeostasis. Hyperactive mTOR signalling in COPD is further augmented by growth factor positive feedback mechanism. Excessive ROS also induces NF-κB/AP-1 activation and chronic inflammation. SIRT6, an antiageing molecule involved in DNA repair and known to inhibit PI3K is reduced in COPD. Excessive DNA damage will result in ATM/p53 activation promoting cell cycle arrest. When cell growth activation via mTOR pathway is combined to cell cycle arrest stimuli, cells activate the senescence pathway. AMPK, 5′-AMP-activated protein kinase; AP, activator protein; ATM, ataxia telangiectasia mutated; FOXO3A, forkhead box O3A; mTOR. mammalian target for rapamycin; NF-κB, nuclear factor-κB; Nrf2, Nuclear factor erythroid 2-related factor 2; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PTEN, phosphatase and tensin homologue; ROS, reactive oxygen species; SIRT, Sirtuin.
Hallmarks of ageing and cellular senescence in COPD
The ageing lung is characterised by loss of elasticity, enlargement of alveoli, low grade inflammation but, in contrast to emphysema, no destruction of alveolar walls.9 Cellular senescence is also present in the lungs of ageing individuals suggesting a predisposition to age-related diseases, such cancer and COPD. Examining and understanding the underlying molecular mechanisms involved in normal lung ageing may help to understand how tobacco smoke and other oxidative stresses may accelerate lung ageing and result in COPD.
The hallmarks of ageing, which include telomere shortening, genomic instability, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, deregulated nutrient-sensing and stem cell exhaustion, are all observed in COPD lungs. As mitochondria are the main source of ROS within the cells, they have been proposed to play a central role in the ageing process.2 In addition to mitochondrial dysfunction, defective ‘retrograde signalling’, caused by impaired signalling between factors reliant on mitochondrial signalling (eg, ROS, ATP) and nuclear genes, can result in impaired formation of new mitochondria (biogenesis) that is also linked to ageing.10 Several studies have shown that mitochondrial metabolism plays a central role in mediating longevity via nutrient-sensing pathways and dietary restriction.10 Insulin/insulin growth factor-1 (IGF-1) signalling and mechanistic target of rapamycin (mTOR) signalling pathways are the two key pathways linked to the regulation of life span.11 Impaired IGF-1 and inhibition of mTOR activity extend life span in worms, flies and mammals.11–13 Activation of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/mTOR pathway limits life span by accelerating age-related diseases.4 mTOR links cellular bioenergetic pathways to longevity and that by decreasing nutrient-sensing pathway longevity can be extended.14 Excessive ROS from cigarette smoke exposure may induce the accelerated cellular senescence that is implicated in the pathogenesis of COPD (figure 1B).8 When antioxidant defences and autophagy are defective and cells are overwhelmed by excessive ROS, the tumour suppressor protein p53 activates the expression of multiple genes that either induce apoptosis or induce transient (quiescence) or permanent (senescence) cell cycle arrest (figure 2).15 Once the cell cycle is blocked, but mTOR signalling is still active, a cell becomes hyperactive and hypertrophic, and compensates by becoming senescent (figure 2).4
PI3K/AKT/mTOR activation in COPD
There is evidence in lung homogenates and peripheral blood mononuclear cells of patients with COPD for activation of the PI3K/AKT axis (figure 2).16 In COPD, the growth factors vascular endothelial growth factor, transforming growth factor-β (TGF-β) and basic fibroblast growth factor are also significantly increased17 and further activate the PI3K/AKT/mTOR pathway, resulting in a positive feedback (figure 2).18 ,19 Furthermore, in vitro data shows that oxidative stress, in the form of hydrogen peroxide (H2O2) or cigarette smoke, activates PI3K and AKT,16 thus indicating that ROS stimulates cell growth and survival pathways. In response to growth stimuli cells have developed mechanisms that counteract the PI3K/AKT/mTOR axis. SH2-containing inositol-5′-phosphatase-1, and phosphatase and tensin homologue (PTEN) are membrane tyrosine phosphatases that reverse PI3K activation.20 These tyrosine phosphatases are oxidation-susceptible, thus excessive ROS reduces their catalytic activity (figure 2).21 Interestingly PTEN polymorphisms are a genetic risk factor for COPD, suggesting that a loss of PTEN could result in the permanent activation of PI3K/AKT observed.22
Epigenetic modifications also play an important role in inhibiting mTOR.20 Sirtuins (SIRTs) are NAD+-dependent histone/protein deacetylases that exhibit differential specificity toward acetylated substrates, thus modulating a range of physiological functions, including gene expression, metabolism and ageing.23 SIRT1 inhibits mTOR via deacetylation and activation of liver kinase B1, an upstream activator of 5′-AMP-activated protein kinase (AMPK).23 In addition, SIRT1 promotes genomic stability.23 SIRT1 is markedly reduced in peripheral blood and lung tissue from patients with COPD due to excessive ROS.24 AMPK also positively regulates SIRT1.25 Another member of the SIRT family, SIRT6, which is also reduced in COPD, functions as a negative regulator of the PI3 K/AKT/mTOR pathway (figure 2).26 SIRT6 is an important modulator of DNA repair, metabolism, telomere structure and nuclear factor-κB (NF-κB).23 SIRT1 and SIRT6 enhance autophagy in mammalian cells, thus playing an important role against senescence.27 ,28 Consequently in COPD, evidence of accelerated ageing due to activated PI3K/AKT/mTOR could be caused by excessive oxidative stress that directly activates and exacerbates PI3K activation, but also through the defective negative feedback mechanism regulated by PTEN, SH2-containing inositol-5′-phosphatase-1, AMPK, SIRT1 and SIRT6, thus resulting in increased production of growth factors and cytokines responsible for chronic inflammation (figure 2). Indeed, sensitivity to oxidative stress is considerably augmented by increased mTOR signalling.29 This is partly due to AKT phosphorylation of the transcription factor forkhead box O3A (FOXO3A), resulting in cytoplasmic retention and subsequently decreased antioxidant response, with decreased expression of key antioxidants such as superoxide dismutases (figure 2).25 FOXO3A expression is significantly decreased in the lungs of smokers and patients with COPD, as well as in those of mice exposed to cigarette smoke.30 The importance of FOXO3 was confirmed by a study in which FOXO3 genetic ablation in mice resulted in cigarette smoke-induced emphysema and an amplified inflammatory response associated with additional oxidative stress due to downregulation of antioxidant genes.30 SIRT1 inactivation also reduces FOXO3A antioxidant response.31 In addition, AKT inactivates histone deacetylase-2, a positive regulator of the antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) and suppressor of inflammatory gene expression.16 ,32 Nrf2 function is reduced in patients with COPD, thus contributing to excessive oxidative stress in the lung (figure 2).33 Additional damage is further compounded by the fact that FOXO3A and SIRT1 function as positive regulators of autophagy and mitochondrial biogenesis, whereas mTOR is a negative regulator.28 ,34 ,35 Excessive ROS also activates redox-sensitive proinflammatory transcription factors, such as NF-κB and activator protein-1, further enhancing inflammation.1 Thus, in addition to activating the PI3K/AKT/mTOR pathway and inhibiting negative feedback mechanisms, ROS are also responsible for the chronic inflammation seen in COPD.1 ROS-induced DNA damage36 and inhibition of SIRT1 and SIRT6, which are important in the maintenance of telomere structure and DNA repair, could tip the balance of PI3K/AKT/mTOR-induced proliferation towards cell cycle arrest. ROS also activate the ataxia telangiectasia mutated/p53 pathway and precipitate cells towards senescence and SASP in COPD (figure 2).37
Autophagy in COPD
Autophagy, in order to keep the interior of a cell clean and fully functional, functions by selectively degrading and removing damaged proteins, organelles and pathogens via lysosomes.38 Ageing cells accumulate damaged and misfolded proteins through a functional decline in autophagy,38 and exposure to cigarette smoke in the elderly can lead to additional cellular damage precipitating cells towards senescence.39 Although autophagy is generally believed to play a protective role in response to exogenous stress, prolonged and excessive autophagy has also been associated with autophagic cell death or ‘autosis’.40 The role of autophagy in bronchial and alveolar epithelial cell COPD could be divided into evidence showing that excessive autophagy or mitophagy results in autophagy mediated cell death against evidence showing that in COPD autophagy is defective.41–45 What is clear is that the markers of autophagy (LC3-II), and mitophagy (PINK1) are increased in lung peripheral tissue from patients with COPD suggesting a possible increase in autophagy or a defect in maturation or resolution.42 ,44 ,46 Choi et al have concluded that excessive autophagy and mitophagy induced by cigarette smoke results in bronchial cell apoptosis and necroptosis, respectively, thus providing a possible mechanism of emphysema.41 ,44 In addition, they found that oxidative stress-mediated increase of histone deacetylase-6 is associated with autophagic degradation and shortening of bronchial cilia suggesting mucociliary dysfunction.43 This is partly supported by a previous study that shows how blockage of autophagy enhances cilia growth.47 In other studies, Kuwano et al found that cigarette smoke transiently activated autophagy in isolated human bronchial epithelial cells from patients with COPD resulting in increased cell senescence and accumulation of autophagic adaptor protein p62 along with ubiquitinated proteins.48 Inhibition of autophagy further increases levels of p62 and ubiquitinated proteins, generating more senescence and SASP with the secretion of interleukin 8 (CXCL8).48 The mTOR inhibitor Torin 1 restores normal autophagic flux by inhibition of mTOR.48 An autophagy-defective Atg5−/− knockout mice also displays increased p62 levels in bronchial epithelial cells which is correlated with a reduction in motile cilia length.49
Differences in the methods for detecting the autophagic flux, together with the type of lung tissue analysed, could explain the contradictory findings. In addition, the association with cell death might be due to the concurrent activation of autophagy and apoptosis, as seen in lung epithelial cells exposed to cigarette smoke. In other cases, autophagic cell death is attributable simply to overwhelming autophagosome formation as a part of the stress response in which cytosol and organelles are destroyed to an unrecoverable degree.50 In fact, inhibition of autophagy increases susceptibility to oxidative damage and apoptosis,38 whereas activation of autophagy in endothelial cells from patients with COPD leads to inhibition of apoptosis.51
The possible role of autophagy in immune cells is less controversial. Macrophages from smokers without COPD display defective autophagic flux resulting in the accumulation of substrate of autophagy, p62 and misfolded proteins due to dysfunctional lysosomal digestion of the autophagosomal load.52 A recent study found that beclin1−/− mice, which are unable to form autophagosomes, are defective in phagocytosis and susceptible to sepsis.53 Such defective phagocytosis resulting in poor clearance of bacteria has been described in alveolar macrophages from patients with COPD.54
Defective mitochondrial function in COPD
Mitochondria can regulate cellular homoeostasis through membrane potential, mitophagy and generation of ROS.10 Ageing is linked to mutations of mitochondrial gene, which encode key proteins of the respiratory chain complex.10 Certain combinations of mitochondrial DNA (mtDNA) mutations (haplogroups) increase, whereas others decrease the risk of COPD. For example, mtDNA haplogroup D, which has Leu→Met substitution at amino acid 237 of mitochondrial Nicotinamide Adenine Dinucleotide plus Hydrogen (NADH) dehydrogenase subunit 2, may protect against oxidative damage.55 Chronic oxidative stress may induce mtDNA damage, resulting in point mutations, insertions and deletions.55 ,56 Oxidative damage accumulation and the subsequent variations in mtDNA sequences generated can eventually result in abnormal oxidative phosphorylation (figure 3).10 The central role that mitochondrial ROS might play in COPD is highlighted by the key role of several antioxidants linked to mitochondrial function in COPD including catalase, heme oxygenase, superoxide dismutase and glutathione.57 Interestingly overexpression of catalase present inside mitochondria extends life span in mice.4 However, ROS production by mitochondria is not the only abnormality linked to cellular dysfunction with ageing and there is mounting evidence suggesting that mitochondrial metabolism is also important in mediating longevity through nutrient-sensing pathway involving insulin/IGF-1 and mTOR signalling pathways.10 Indeed increased ROS may enhance this signalling, as discussed above.

{kind=link}
{kind=link}
{kind=link}
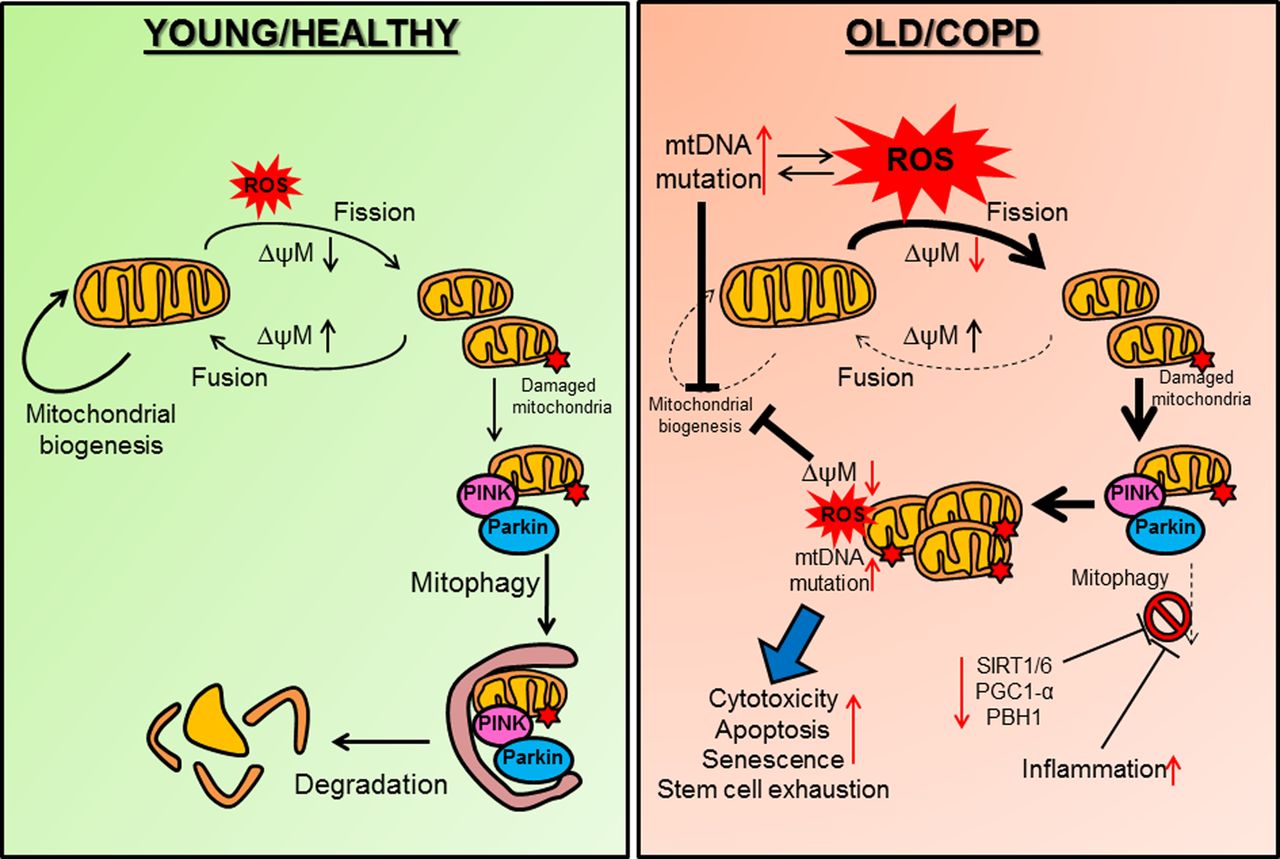
Defective mitochondrial function in COPD. (Left) In healthy individuals exposure to environmental or exogenous oxidants (represented by ROS) induces a decrease in mitochondrial potential (ΔψM) resulting in mitochondrial fission. If the damage to mitochondria is reversible, an increase in Δψm will lead to mitochondrial fusion. Damaged mitochondria are targeted for mitophagy (degradation) when PINK1 accumulates on the outer mitochondrial membrane and binds to Parkin. An effective mitochondrial biogenesis is important for mitochondria integrity against ROS. (Right) In COPD, mitochondrial biogenesis is reduced thus favouring ROS-induced mitochondrial fission and damage. Excessive ROS, which could also result from defective mitochondria induces mtDNA mutations and impairs mitochondrial biogenesis. Damaged mitochondria are not cleared properly by mitophagy as important molecules that regulate mitophagy are decreased in COPD (SIRT1, SIRT6, PGC-1α and PBH1). Inflammation can also impair mitophagy. Accumulation of damaged mitochondria will further affect mitochondrial biogenesis in a positive feedback loop by generating additional ROS and mtDNA mutations. This leads to increased cell cytotoxicity, apoptosis, senescence and stem cell exhaustion. mtDNA, mitochondrial DNA; PBH1, prohibitin 1; PGC1-α, peroxisome proliferator-activated receptor-γ coactivator 1α; PINK1, phosphatase and tensin homologue (PTEN) induced putative kinase 1; ROS, reactive oxygen species; SIRT, Sirtuin.
Prohibitin-1 (PHB1), a mitochondrial protein that interacts with the NADH dehydrogenase protein complex, is significantly downregulated in bronchial epithelial cells from patients with COPD.58 PBH1 plays a crucial role in preserving normal mitochondrial function and morphology and is known to be linked to ageing and autophagy.58 Decreased PHB1 levels together with impaired autophagy increases the susceptibility of cells to mitochondrial damage and death.59 PHB1 is also highly expressed in the vascular system and ablation of PHB1 in endothelial cells increases mitochondrial ROS production and activates PI3K/mTOR signalling, leading to cellular senescence.60
Impairment in mitochondrial turnover is an additional mechanism that links mitochondrial dysfunction to inflammation.61 Mitochondria fuse in order to maintain intact mitochondrial DNA copies, mitochondrial membrane components and matrix metabolites.61 If mitochondrial fusion or fission events are defective this results in defective oxidative phosphorylation deficiencies, the loss of mtDNA and exaggerated generation of ROS.56 ,61 When the mitochondrial membrane potential (Δψm) is low, it leads to mitochondrial fission and induction of mitophagy in daughter mitochondria (figure 3). In fact, mitochondrial fission is predominant in diseased cells where the elimination of damaged mitochondria takes place via mitophagy.62 In bronchial epithelial cells from patients with COPD the mitochondria appear fragmented.44 ,63 ROS from cigarette smoke and endogenous ROS induce mitochondrial fragmentation in primary human bronchial epithelial cells, with subsequent additional production of ROS from fragmented mitochondria mainly resulting in acceleration of cellular senescence.46 ,64 Furthermore, long-term cigarette smoke exposure increases the expression of fission/fusion markers, oxidative phosphorylation proteins and markers of oxidative stress such as manganese superoxide dismutase (Mn-SOD).44 ,46 In COPD airway epithelial cells these changes are associated with increased secretion of proinflammatory mediators, such as CXCL8, interleukin (IL)-6 and IL-1β.46
Mitochondrial clearance is closely linked with mitochondrial fission and fusion proteins.62 Recent studies suggest that E3 ubiquitin ligase, Parkin/PARK2 and PTEN-induced putative protein kinase 1 (PINK1) are the main regulators in the elimination of dysfunctional mitochondria.65 ,66 PINK1 is usually found at low levels on mitochondria, but when the Δψm is reduced due to stress, PINK1 accumulates on the outer mitochondrial membrane where it binds Parkin.67 The PINK1/Parkin complex isolates dysfunctional from healthy mitochondria directing them towards elimination by mitophagy (figure 3).61 In this way mitophagy is able to control excessive mitochondrial ROS from degrading damaged mitochondria.61 Mitophagy is induced by moderate levels of ROS in a mitochondrial fission-dependent manner.68 PINK1 expression is increased in COPD suggesting that initiation of mitophagy might be taking place, but there is no evidence of resolution.46 A recent study confirmed that PINK1 is increased in COPD resulting in excessive mitophagy responsible for necroptosis of bronchial and alveolar epithelial cells.44 The same study showed that PINK1−/− (but not Parkin−/−) mice are protected against cigarette smoke-induced mitochondrial dysfunction.44 The deleterious role of mitophagy in COPD is challenged by recent findings in which autophagy-defective Atg5−/− aged mice displayed defective mitochondria in bronchial epithelial cells that were associated with altered lung mechanics.49 Additional evidence suggests that defective mitophagy might be detrimental as in macrophages from smokers mitophagy is impaired due to a failure in the autophagic flux (figure 3).52 SIRT1 positively modulates mitophagy through the upregulation of the coactivator peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α).69 The expression of PGC-1α is reduced in the lungs of patients with moderate and severe COPD.70 Also, elevated levels of Tumor Necrosis Factor alpha (TNF-α), which are high in COPD lungs, and which systemically decrease PGC-1α mRNA, indicate suppressed genesis of mitochondria.71 Inflammation inhibits mitophagy, which in turn could amplify mitochondrial dysfunction (figure 3).72 The role of mitophagy in COPD should be further explored as it could help explain why, despite smoking cessation, patients with COPD have fragmented mitochondria, and also why ROS generation persists.
Stem cell exhaustion and senescence in COPD
At a young age, senescence is an advantageous compensatory response that allows the clearance of damaged and potentially oncogenic cells.6 However, this requires an effective cell replacement system and the mobilisation of progenitor cells to establish cell numbers again (figure 1A).14 In aged organisms, this repair system may become inefficient due to the impaired regenerative capacity of stem cells, leading to the accumulation of senescent cells that may further exacerbate the damage (figure 1B).14 In emphysematous lungs, there is considerable evidence for senescence, such as decreased telomere length and enhanced expression of p21, p16, p19 and β-galactosidase activity.8 ,73
In COPD parenchymal destruction and progressive dilation of alveolar spaces is linked to the breakdown of extracellular proteins, such as elastin, and loss of epithelial and endothelial cells of the alveolar wall.8 Damaged alveolar cells can be replaced by migrating progenitor cells but depletion of these cells could result in impaired ability to repair tissue.74 Senescence of mesenchymal progenitor cells decreases regenerative potential, thus limiting the ability of the lung to repair in response to injury.75 Senescence in mesenchymal stem cells (fibroblast, endothelial cells) could be a causative mechanism of emphysema.8 The size of stem cell populations depends on the balance between self-renewal and cell differentiation.75 When the rate of self-renewal is superior than that of differentiation, the stem cell population increases, whereas when the self-renewal rate is lower than the rate of differentiation, the population declines due to exhaustion (figure 1B).75 In stem cells, ROS forces cells out of quiescence and into proliferation by activating PI3K/AKT and further promoting the production of ROS, thus repressing FOXO-mediated stress response and autophagy.75 Stem cell depletion may be important in COPD as persistent oxidative stress could force stem cells out of quiescence (figure 1A). Alveolar type II (AT-II) cells, which may be the progenitors of type I cells (AT-I) show senescence in COPD.76 ,77 AT-II cells are capable of self-renewal and normally become activated by hyperoxic injury or damage to AT-I cells resulting in re-epithelialisation of the lung.78 A transmembrane tyrosine kinase receptor (c-kit) binds a stem cell factor thus promoting cell proliferation and differentiation.79 Lindsey et al80 showed that c-kit ablation in mice resulted in spontaneous airspace enlargement similar to emphysema, which suggested a possible role in COPD. Recent evidence from circulating endothelial progenitor cells (blood outgrowth endothelial cells) from smokers and patients with COPD shows increased DNA double-strand breaks associated with senescence and impaired capacity for vascular repair compared with non-smokers.37 Also, senescence is negatively associated with SIRT1 activity and expression.37 Exhaustion and senescence of endothelial progenitor cells provides a link between COPD and cardiovascular disease, a common comorbidity associated with COPD.
Immunosenescence in COPD
During ageing the immune system loses its efficacy, resulting in increased vulnerability to infectious diseases and to pathological conditions relating to inflammation (eg, Alzheimer’s disease, cardiovascular disease) or autoimmunity (eg, rheumatoid arthritis).81 This overall loss in immune competence is termed ‘immunosenescence’.81 Increased PI3K/AKT/mTOR signalling is found in autoimmune lymphoproliferative disease and inhibition of PI3K can prevent autoimmunity, suggesting a central role for mTOR signalling in immunosenescence.82
Age-associated alterations in Toll-like receptor function in human dendritic cells are associated with decreased T cell-mediated innate immunity, resulting in reduced ability to fight pathogens in the elderly.83 Peripheral blood monocytic cells and circulating leucocytes of patients with COPD show evidence of T cell senescence and autoimmunity.84 Increased CD4+CD28null T cells in COPD indicate that chronic exposure to cigarette smoke can result in the loss of CD28 and increase of natural killer cell receptor expression on T cells.85 Multiple host defence mechanisms, such as those provided by the airway epithelial barrier and innate immune cells, including alveolar macrophages, neutrophils, dendritic cells and natural killer cells, are broadly suppressed in COPD.86 A recent study found that higher levels of T helper 17 cells, but lower levels of T regulator cell (Treg) in circulating T cells were associated with disease progression and impaired lung function in patients with COPD, thus linking COPD and autoimmunity.87 Immunosenescence contributes to an increased susceptibility of the elderly to malignancies and the suppression of the innate immune mechanisms in patients with COPD may explain why COPD is associated with a marked increase in risk of developing lung cancer.86
Concluding remarks and therapeutic prospects
Long-term cigarette smoke exposure can progressively deplete cells of their antioxidant and autophagic defences, reducing antiageing molecules, impairing DNA repair process and mitochondrial dysfunction, thereby driving cells towards apoptosis, senescence or stem cell exhaustion. Increasing evidence in COPD suggests that ageing of the lung is accelerated; the result of hypertrophic and hyperactive cells caused by excessive oxidative stress and deregulated mTOR signalling. This leads to chronic inflammation or SASP which increases the risk of comorbidities such as cancer, atherosclerosis, diabetes and metabolic disease.
Identification of the signalling pathways that are involved in accelerated ageing in COPD has led to the discovery of novel therapeutic targets and so may lead to new treatments. Antiageing therapies that target the accelerated ageing process hold promise for the long-term treatment of ageing-related diseases providing a good therapeutic ratio can be established and this may be favoured in the treatment of COPD by inhaled delivery of drugs to reduce systemic side effects that might be otherwise dose-limiting. Some drugs, termed ‘geroprotectors’, have already been identified, and may be a starting point to the discovery of safe therapies (figure 2).25 Reducing the oxidative stress damage by reversing defective antioxidant response or inhibiting the mTOR pathway with rapamycin or metformin could prevent senescence and chronic inflammation. Importantly, geroprotectors are not conceived as therapies to extend life span but to reduce acceleration of the ageing process or prevent disruption of the repair mechanism due to oxidative stress and cigarette smoking. There is now an active search for more effective antioxidants and particular interest in developing drugs that will activate or restore the activity of Nrf2, which is impaired in COPD cells. Drugs that interfere with mTOR signalling, such as rapamycin, are already in clinical use, although there is an unacceptable level of toxicity. Metformin, which interacts with this pathway, is better tolerated and in widespread use to treat type-2 diabetes.88 Spermidine, a naturally occurring polyamine can restore autophagy activity and reduce oxidative stress.88 Low dose theophylline, which is well tolerated, may also be effective and is currently in clinical trials for COPD. Future effort should be focused on either preventing or protecting cells against senescence and SASP. Restoring exhausted stem cells using stem cell therapies was recently achieved by reprogramming COPD fibroblast through the induction of pluripotent stem cells.89
References
Footnotes
-
Contributors NM wrote the manuscript. KI and PJB participated in revising the manuscript by giving critically important intellectual content.
-
Funding Wellcome Trust Programme Grant (093080/Z/10/Z) and NIHR Respiratory Biomedical Research Unit.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.