Article Text

Abstract
Background Steroid-insensitive endotypes of asthma are an important clinical problem and effective therapies are required. They are associated with bacterial infection and non-eosinophilic inflammatory responses in the asthmatic lung. Macrolide therapy is effective in steroid-insensitive endotypes, such as non-eosinophilic asthma. However, whether the effects of macrolides are due to antimicrobial or anti-inflammatory mechanisms is not known.
Objective To determine and assess the efficacy of macrolide (ie, clarithromycin) and non-macrolide (ie, amoxicillin) antibiotic treatments in experimental models of infection-induced, severe, steroid-insensitive neutrophilic allergic airways disease (SSIAAD), compared with steroid-sensitive AAD and to delineate the antimicrobial and anti-inflammatory effects of macrolide therapy.
Methods We developed and used novel mouse models of Chlamydia and Haemophilus lung infection-induced SSIAAD. We used these models to investigate the effects of clarithromycin and amoxicillin treatment on immune responses and airways hyper-responsiveness (AHR) in Ova-induced, T helper lymphocyte (Th) 2 -associated steroid-sensitive AAD and infection-induced Th1/Th17-associated SSIAAD compared with dexamethasone treatment.
Results Clarithromycin and amoxicillin had similar antimicrobial effects on infection. Amoxicillin did attenuate some features, but did not broadly suppress either form of AAD. It did restore steroid sensitivity in SSIAAD by reducing infection. In contrast, clarithromycin alone widely suppressed inflammation and AHR in both steroid-sensitive AAD and SSIAAD. This occurred through reductions in Th2 responses that drive steroid-sensitive eosinophilic AAD and tumour necrosis factor α and interleukin 17 responses that induce SSIAAD.
Conclusions Macrolides have broad anti-inflammatory effects in AAD that are likely independent of their antimicrobial effects. The specific responses that are suppressed are dependent upon the responses that dominate during AAD.
- Asthma
- Respiratory Infection
- Allergic lung disease
- Bacterial Infection
Statistics from Altmetric.com
Key messages
What is the key question?
-
Can macrolides be used to treat infection-induced steroid-insensitive severe asthma and how does this work?
What is the bottom line?
-
Clarithromycin suppresses severe, steroid-insensitive allergic airways disease through its anti-inflammatory effects on tumour necrosis factor-α/interleukin-17 immune responses that are largely independent of its antimicrobial effects.
Why read on?
-
This study demonstrates how macrolide therapies may be effective in infection-induced steroid-insensitive asthma.
Introduction
Inhaled corticosteroids are the mainstay therapy for the management of asthma. However, they only suppress the symptoms and not the underlying causes of the disease. Significantly, between 5% and 10% of asthmatics are refractory to steroid treatment,1 and this group accounts for >50% of asthma-associated healthcare costs.2 Steroid-insensitive asthmatics typically have more severe disease, which is commonly characterised by non-eosinophilic or neutrophilic airway inflammation.3 Effective therapies are urgently required for steroid-insensitive asthma. However, the limited understanding of the mechanisms and aetiological factors that underpin steroid-insensitivity has restricted the development of such treatments.
Clinical studies show associations between increases in the expression of T helper lymphocyte type (Th)1 and Th17 cytokines with neutrophilic inflammation and severe, steroid-insensitive (SSI) asthma.2 ,4 Experimental studies show that adoptive transfer of Th1 and Th17 cells induce a phenotype of allergic airways disease (AAD) in mice that is characterised by increased neutrophilic airway inflammation and is more resistant to steroid treatment than Th2 cell-induced AAD.5 ,6 These studies suggest that Th1 and/or Th17 immune responses in the lung may result in more severe endotypes of asthma that are steroid-insensitive.
Numerous clinical studies have linked Chlamydia pneumoniae (Cpn) and Haemophilus influenzae (Hi) infections to steroid-insensitive endotypes such as neutrophilic asthma. Cpn lung infection induces strong neutrophilic inflammation and potent Th1 and Th17 responses, which are required to clear the infection.7 ,8 Furthermore, increased airway neutrophil numbers in SSI asthmatics predict the presence of Chlamydia.9 Hi infections also induce neutrophilic inflammation and Th17 responses.10 ,11 One study showed that in neutrophilic asthmatics, 43% were colonised by bacteria, and Hi was most commonly isolated.12 We previously showed that neutrophilic asthmatics had significantly higher loads of bacteria. Hi was detected in 60% of these patients, who were taking higher doses of steroids.13 Together, these studies provide strong associations between infections that induce Th1/Th17 responses and neutrophilic inflammation with steroid-insensitivity in severe asthma.
Specific treatments for infection-induced SSI asthma are not widely used and alternative approaches are needed. One strategy is the use of infection- and/or immune-targeted therapies such as antibiotics, particularly macrolides. Some antibiotics, such as β-lactams like amoxicillin, have antimicrobial but not broad anti-inflammatory properties. Macrolides are potent antimicrobial agents and first-line treatments for some bacterial infections. They inhibit bacterial protein synthesis, adherence, motility and biofilm formation.14 ,15 Macrolides also have immunomodulatory properties, and in severe asthma, clarithromycin suppresses airway inflammatory cell infiltration and airway hyper-responsiveness (AHR) and enhances steroid responses.16 ,17 Importantly, however, it is unknown whether the effects of macrolides are underpinned by their antimicrobial or anti-inflammatory properties. Elucidating the effects of macrolides may highlight their potential use and further the understanding of how we may optimally treat SSI asthma.
Here, we developed a novel mouse model of Chlamydia infection-induced, severe, steroid-insensitive, neutrophilic AAD (SSIAAD). We used it as well as a model of Hi-induced SSIAAD we previously developed18 to compare the efficacy of non-macrolide and macrolide antibiotics (amoxicillin and clarithromycin) that are in widespread clinical use, in treating this endotype. We show that clarithromycin, but not amoxicillin, is an effective treatment for steroid-sensitive AAD and SSIAAD even in the absence of steroids, and efficacy results from anti-inflammatory rather than antimicrobial properties.
Methods
See online supplement for additional details.
Experimental models
In our new model of SSIAAD, female (6–8 weeks old) BALB/c mice were intraperitoneally (IP) sensitised to ovalbumin (Ova, 50 μg (Sigma-Aldrich, Castle Hill, Australia)), with the adjuvant alum (1 mg Rehydrogel (Reheis, Berkeley Heights, New Jersey, USA) in 200 μL 0.9% saline) on day 0.19 Mice were then intranasally (IN) challenged with Ova on d12–13 and d33–34 (10 μg; 50 μL sterile saline (figure 1A)). On d14, mice were inoculated IN with the natural mouse pathogen Chlamydia muridarum (Cmu; 100 inclusion-forming units, ATCCVR-123, 30 μL sucrose phosphate glutamate buffer (SPG)).19 ,20 Dexamethasone (DEX) was administered IN (2 mg/kg; 50 μL phosphate buffered saline (PBS)) on d32–34 with Ova challenges (figure 1A). We investigated the broader applicability of our data in another established model using Hi infection. Mice were inoculated intratracheally (IT) with 5×105 colony-forming units of non-typeable Hi (NTHi-289, 30 μL PBS) 10d prior (d-10) to Ova IP sensitisation and challenged on d12–15 (figure 1B). Mice received DEX (1 mg/kg; 50 μL PBS) IN on d13–15.10 Controls were sham-sensitised with saline, and sham-inoculated with SPG (Chlamydia-induced AAD) or PBS (Hi-induced AAD). Mice were sacrificed 24 h after the final challenge and features of AAD were assessed.
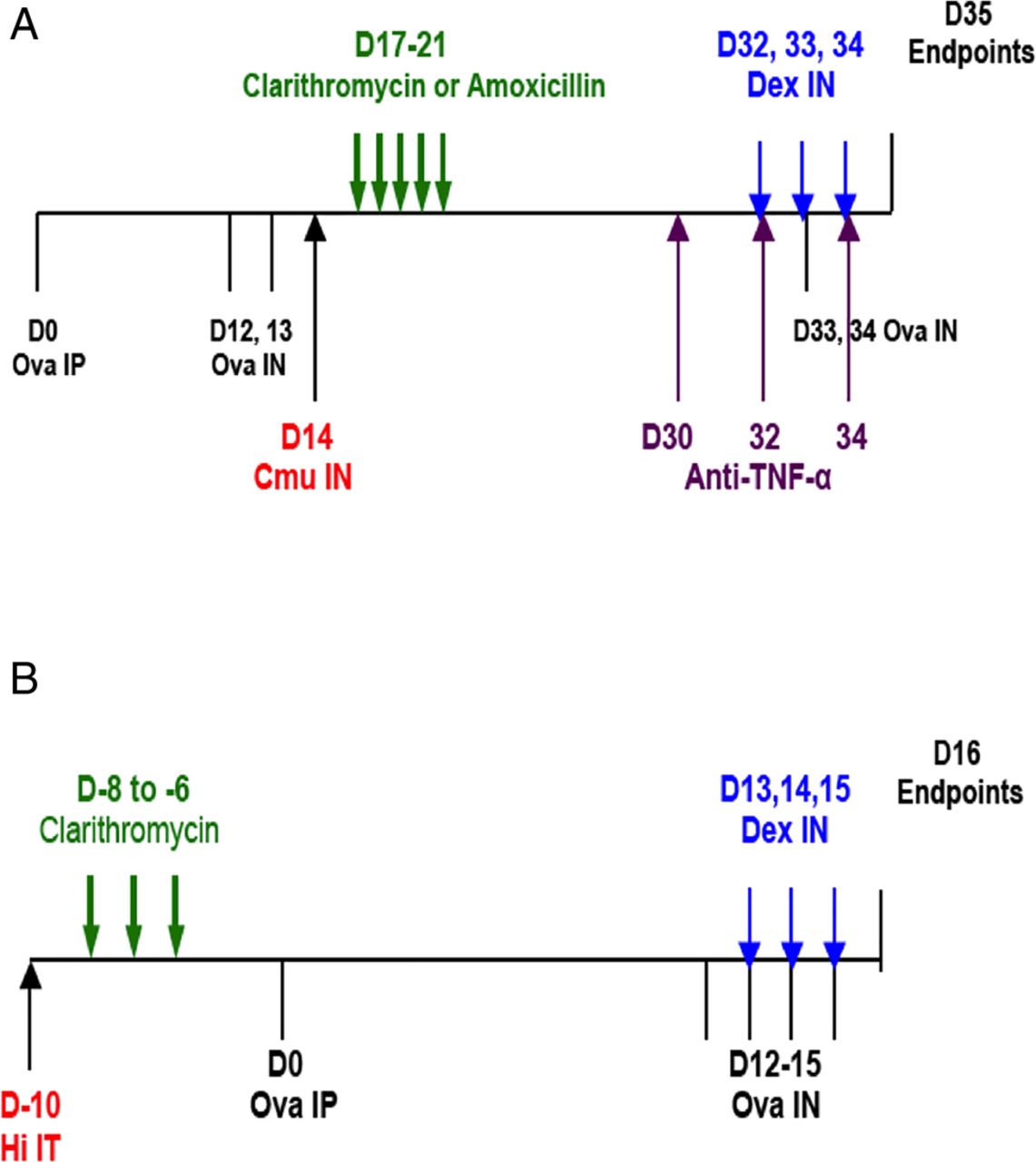
Novel models of infection-induced, severe, steroid-insensitive, neutrophilic allergic airways disease (SSIAAD). Mice were sensitised to ovalbumin (Ova) by intraperitoneal (IP) injection (d0) and AAD was induced by intranasal (IN) Ova challenge (d12–13) followed by rechallenge (d33–34, A). Infected groups were inoculated IN with 100 inclusion-forming units of Chlamydia muridarum in between the two sets of Ova challenges (Cmu; d14). Antibiotics, dexamethasone (DEX) and anti-tumour necrosis factor α (anti-TNF-α) treatments were administered by gavage (d17–21) and IN (DEX d32–34 and anti-TNF-α d30, 32, 34), respectively. Uninfected controls were sham-inoculated with sucrose phosphate glutamate, and non-allergic controls were sham-sensitised with saline (Sal). Mice were sensitised to Ova IP (d0) and AAD induced by IN Ova challenge (d12–15, B). Infected groups were inoculated with 5×105 colony forming units of Haemophilus influenzae (Hi) intratracheally (IT; d-10). Antibiotic and DEX treatments were administered d-8 to d-6 and d13–15, respectively. Uninfected controls received phosphate buffered saline, and non-allergic controls were sham-sensitised with Sal. All features of AAD were assessed 24 h after the final Ova challenge.
Antibiotic treatment
Amoxicillin:clavulanate or clarithromycin (5 mg/kg in 200 μL PBS) was administered by oral gavage on d17–21 (Chlamydia-induced SSIAAD (figure 1A)) or d-8 to d-6 (Hi-induced SSIAAD (figure 1B)). Controls received PBS.
Anti-TNF-α therapy
Anti-tumour necrosis factor α (Anti-TNF- α; 50 μg, 50 μL PBS, (BioXCell, West Lebanon, USA)) monoclonal antibody was administered IN to Chlamydia-infected groups with AAD (Ova/Cmu/αTNF-α) on d30, 32 and 34.
Airway inflammation
Differential leucocyte counts were obtained from May-Grunwald Giemsa stained bronchoalveolar lavage fluid (BALF) cells, using light microscopy.10
Lung function
AHR was measured by anaesthetised, cannulated mice using the Scireq flexiVent FX1 system (Montreal, Canada) for all Chlamydia experiments, or Buxco Electronic resistance and compliance system (Sharon, Connecticut, USA) for all Hi experiments.10 ,19 Data are represented as airways resistance at 10 mg/kg methacholine (figures 1–7) and as dose responsive curves (see online supplement).
Lung mRNA expression
RNA was extracted from homogenised whole lung tissue and reverse transcribed. Relative abundance of cytokine complementary DNA was determined compared to the reference gene hypoxanthine-guanine phosphoribosyltransferase by real-time quantitative PCR.20 ,21 We previously showed that lung cytokine messenger RNA (mRNA) levels correlate with protein levels.22
ELISA
Concentrations of cytokines were determined in Ova- (interleukin (IL)-5, IL-13 and TNF-α) or Hi- (IL-17) stimulated mediastinal lymph node (MLN) culture supernatants by ELISA (R&D systems, Minnesota, USA).18
Statistics
Data are represented as mean±SEM with 6–12 mice in each group. Statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey's or Fisher's least significant difference post-tests. AHR was analysed using two-way repeated measures ANOVA with Tukey's post-test. (GraphPad Prism V.6 Software, San Diego, California, USA). See online supplement for tables of full statistical comparisons of all groups.
Results
Infection-induced SSIAAD
To investigate the effects of antibiotics, we developed a novel model of Chlamydia-induced SSI asthma, and compared it with our model of Hi-induced SSI asthma (figure 1). Treatment groups received antibiotics on d17–21 (3–7 days post infection), and/or DEX on d32–34 (figure 1A, Chlamydia), or antibiotics on d-8 to d-6 and/or DEX on d13–15 (figure 1B, Hi).
Ova-induced AAD (Ova groups) was characterised by increased airway inflammation, Th2 responses and AHR compared with non-allergic (saline) controls (figures 2⇓⇓⇓⇓–7). DEX alone treatment significantly reduced all these cardinal features of AAD. Chlamydia infection (Ova/Cmu), however, increased airway neutrophils and TNF-α responses and decreased eosinophils and Th2 responses compared with uninfected controls (Ova). Inflammation and AHR were insensitive to DEX treatment.

Amoxicillin treatment suppresses some features of steroid-sensitive allergic airways disease and clarithromycin suppresses all features of disease. Effects of amoxicillin and clarithromycin treatment on total cells (A), neutrophils (B), eosinophils (C) and macrophages (D) in bronchoalveolar lavage fluid of steroid-sensitive groups were assessed. Airways hyper-responsiveness in terms of airways resistance at the maximal dose of methacholine (10 mg/mL, E) was also assessed. Results are representative of two independent experiments, with a total of five to eight mice/group and all data are presented as means±SEM. ++++p<0.0001, +++p<0.001 compared with non-allergic (Sal) controls, ####p<0.0001, ###p<0.001, ##p<0.01, #p<0.05 compared with allergic (Ova) controls.

Clarithromycin, but not amoxicillin, treatment broadly suppresses Chlamydia-induced, severe, steroid-insensitive neutrophilic allergic airways disease (SSIAAD). Effects of amoxicillin and clarithromycin treatment on total cells (A), neutrophils (B), eosinophils (C) and macrophages (D) in bronchoalveolar lavage fluid of SSIAAD groups were assessed. Airways hyper-responsiveness in terms of airways resistance at the maximal dose of methacholine (10 mg/mL, E) and Chlamydia 16S RNA levels in the lungs (F) were also assessed. Results are representative of two independent experiments, with a total of five to eight mice/group, and all data are presented as means±SEM. ++++p<0.0001, +++p<0.001 compared with non-allergic (Sal/Cmu) controls, ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05 compared with infected allergic (Ova/Cmu) groups.

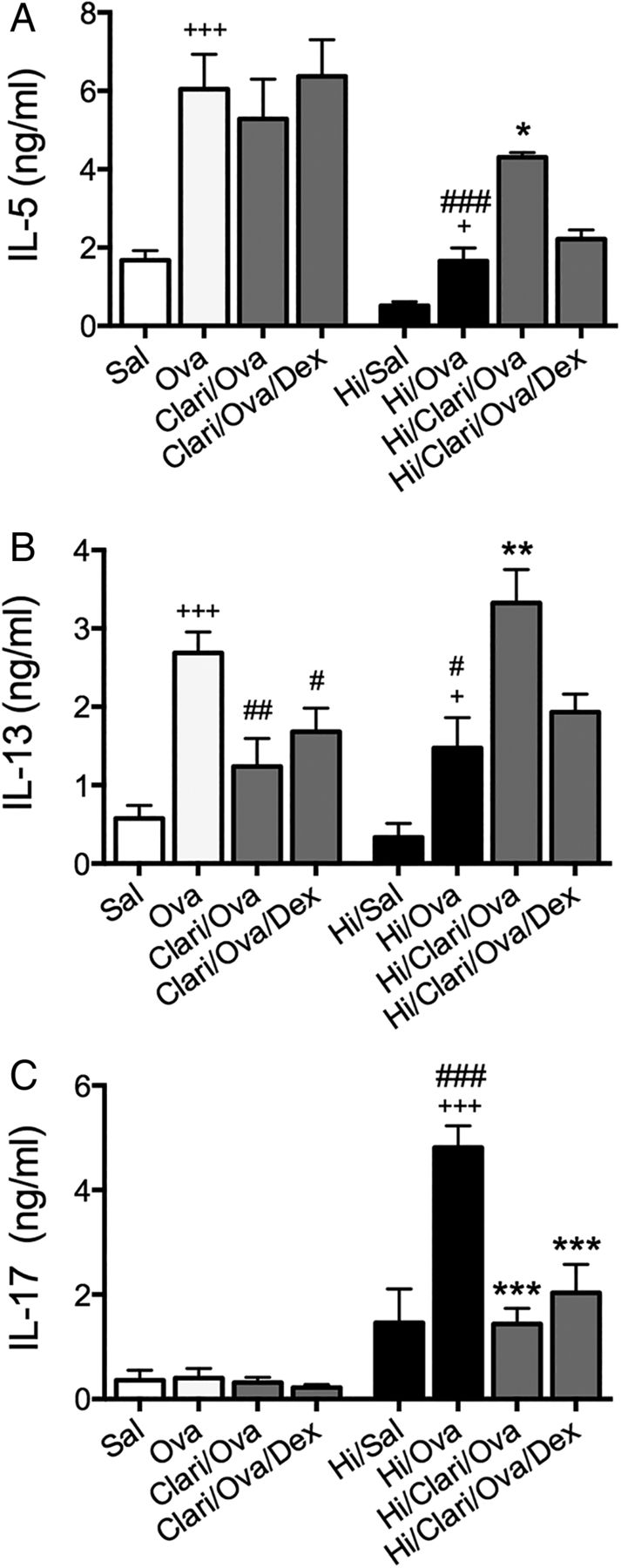
Clathrithromycin, but not amoxicillin, treatment broadly suppresses important inflammatory mediators in steroid-sensitive and Chlamydia-induced, severe, steroid-insensitive, neutrophilic allergic airways disease (SSIAAD). Effects of amoxicillin and clarithromycin treatment on interleukin (IL)-5 (A), IL-13 (B) and tumour necrosis factor α (TNF-α; C) protein release from mediastinal lymph nodes in steroid-sensitive groups, and IL-5 (D), IL-13 (E) TNF-α (F) protein in SSIAAD groups were assessed. Effects of treatments on lung mRNA expression of IL-5, IL-13 and TNF-α in steroid-sensitive (G–I) and SSIAAD groups (J–L) were also assessed. Results are representative of two independent experiments, with a total of 5–10 mice/group, and all data are presented as means±SEM. ++++p<0.0001, +++p<0.001, ++p<0.01, +p<0.05 compared with non-allergic (Sal, or Sal/Cmu) controls, ####p<0.0001, ###p<0.001, ##p<0.01, #p<0.05 compared with allergic (Ova) controls, ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05 compared with infected, allergic (Ova/Cmu) groups.
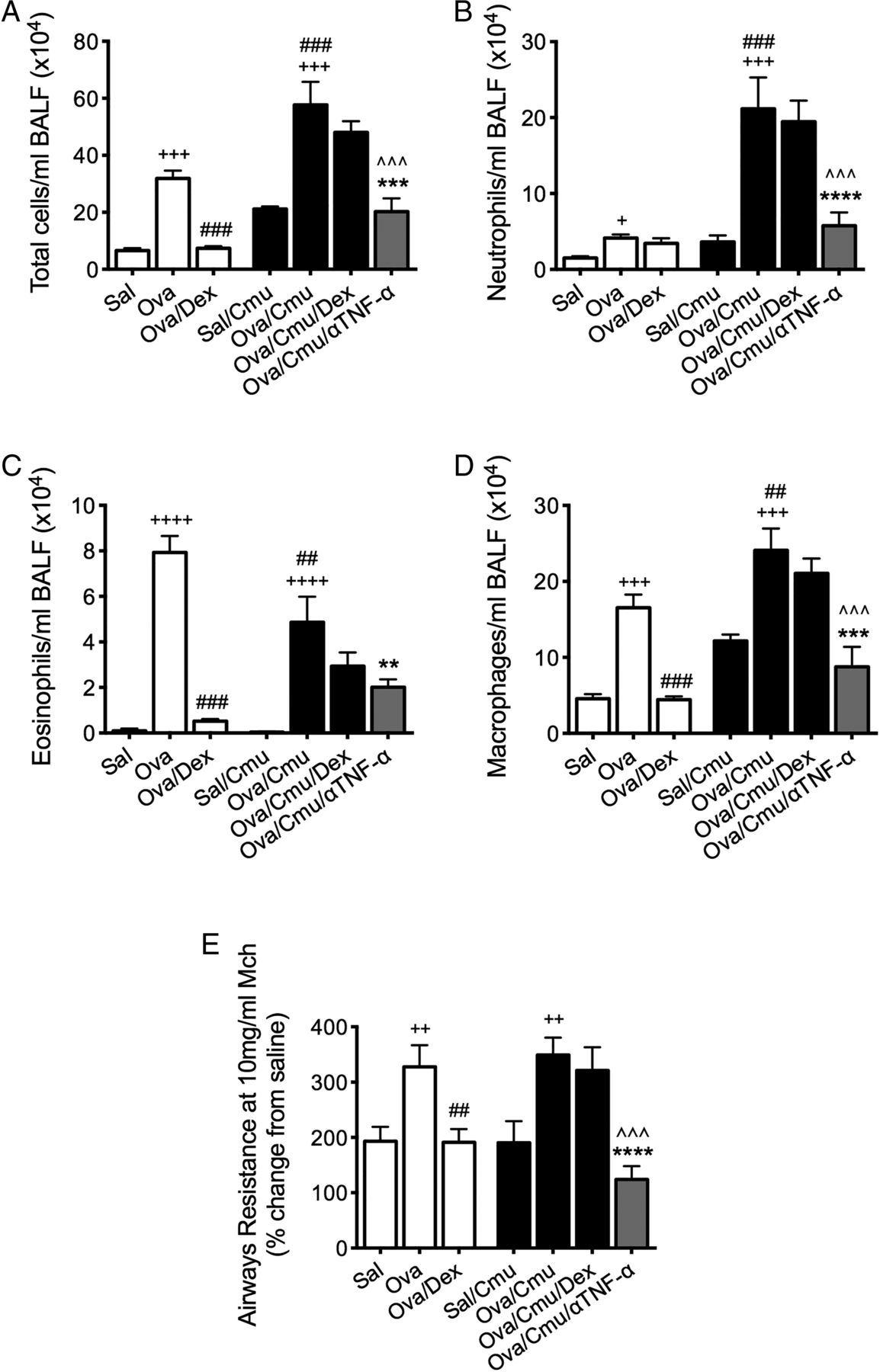
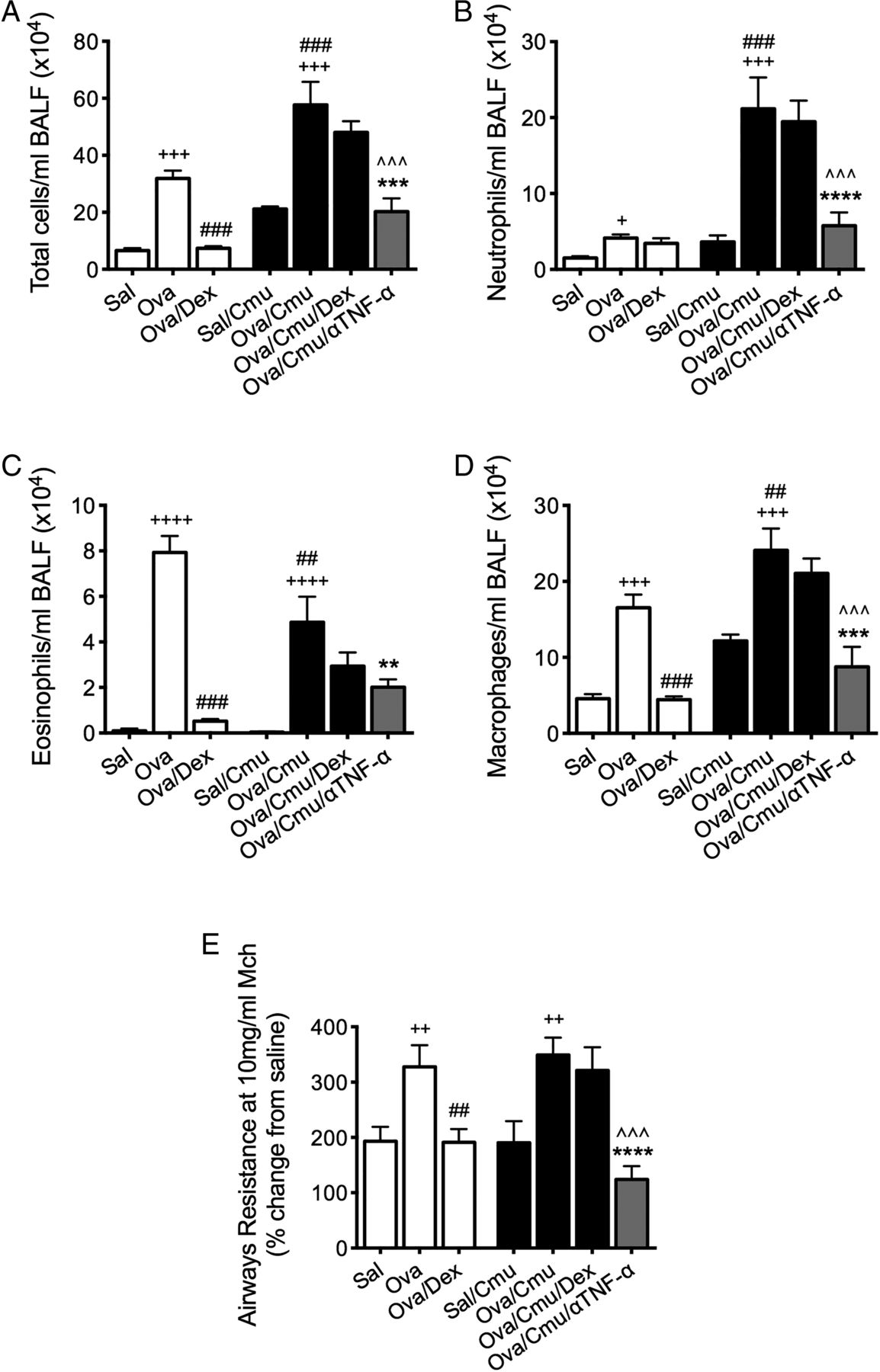
Anti-tumour necrosis factor α (anti-TNF-α) therapy suppresses Chlamydia-induced, severe, steroid-insensitive, neutrophilic allergic airways disease. Effects of anti-TNF-α treatment on total cells (A), neutrophils (B), eosinophils (C) and macrophages (D) in bronchoalveolar lavage fluid (BALF). Airways hyper-responsiveness in terms of airways resistance at the maximal dose of methacholine (10 mg/mL, E) was also assessed. Results are representative of one experiment with 4–10 mice per group, and all data are presented as means±SEM. +++p<0.001, ++p<0.01, +p<0.05 compared with non-allergic (Sal, or Sal/Cmu) controls, ###p<0.001, ##p<0.01, #p<0.05 compared with allergic (Ova) controls, ***p<0.001, **p<0.01, *p<0.05 compared with infected, allergic (Ova/Cmu) groups, ^^^p<0.001, ^p<0.05 compared with infected, allergic, dexamethasone (DEX)-treated (Ova/Cmu/Dex) groups.

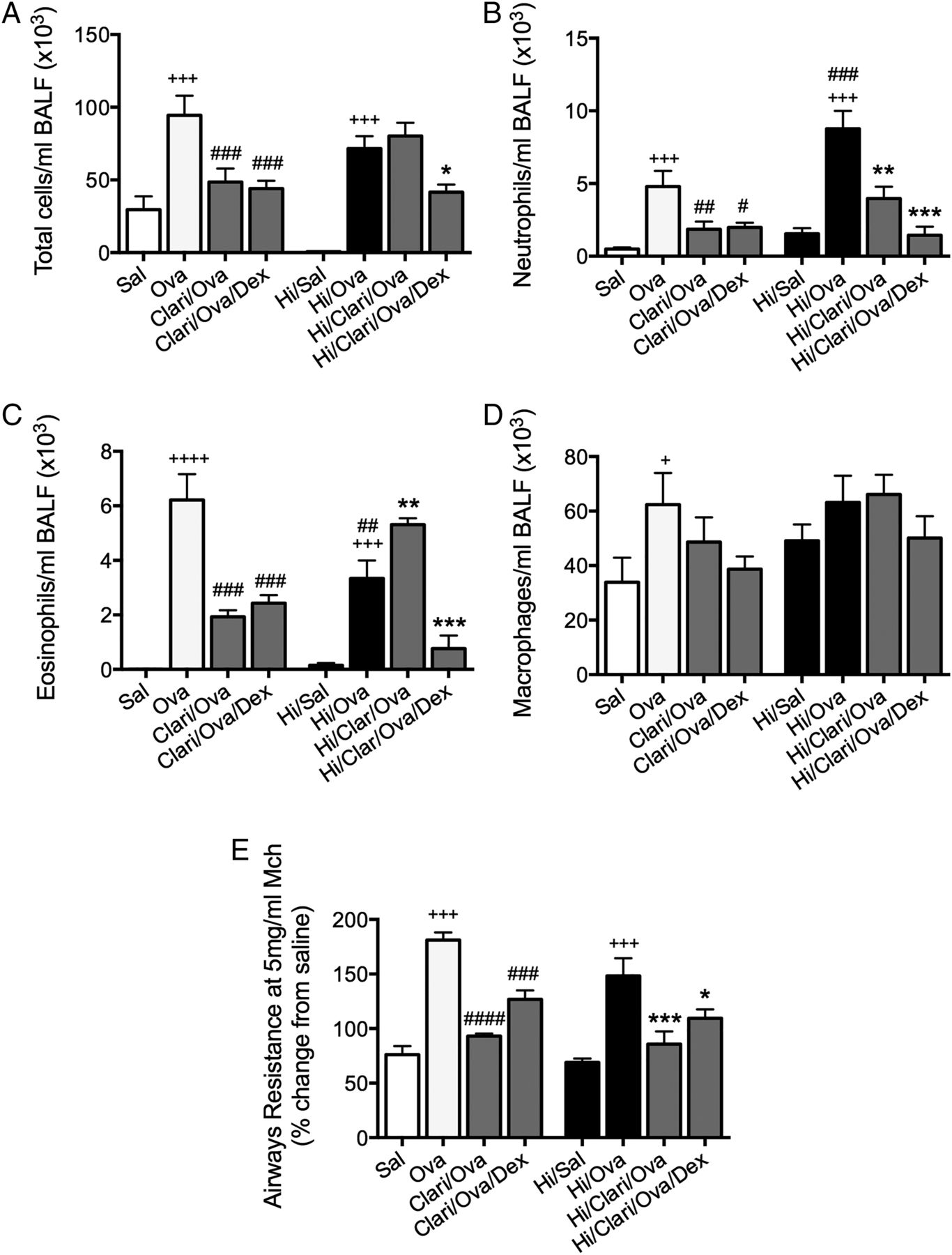
Clarithromycin suppresses Haemophilus influenzae (Hi)-induced, severe, steroid-insensitive, neutrophilic allergic airways disease. Effects of clarithromycin treatment on total cells (A), neutrophils (B), eosinophils (C) and macrophages (D) in bronchoalveolar lavage fluid(BALF). Airways hyper-responsiveness in terms of airways resistance at the maximal dose of methacholine (5 mg/mL, E) was also assessed. Results are representative of two independent experiments, with a total of six mice/group, and all data are presented as means±SEM. ++++p<0.0001, +++p<0.001, +p<0.05 compared with non-allergic (Sal, or Hi/Sal) controls, ####p<0.0001, ###p<0.001, ##p<0.01, #p<0.05 compared with allergic (Ova) controls, ***p<0.001, **p<0.01, *p<0.05 compared with infected, allergic (Hi/Ova) groups.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
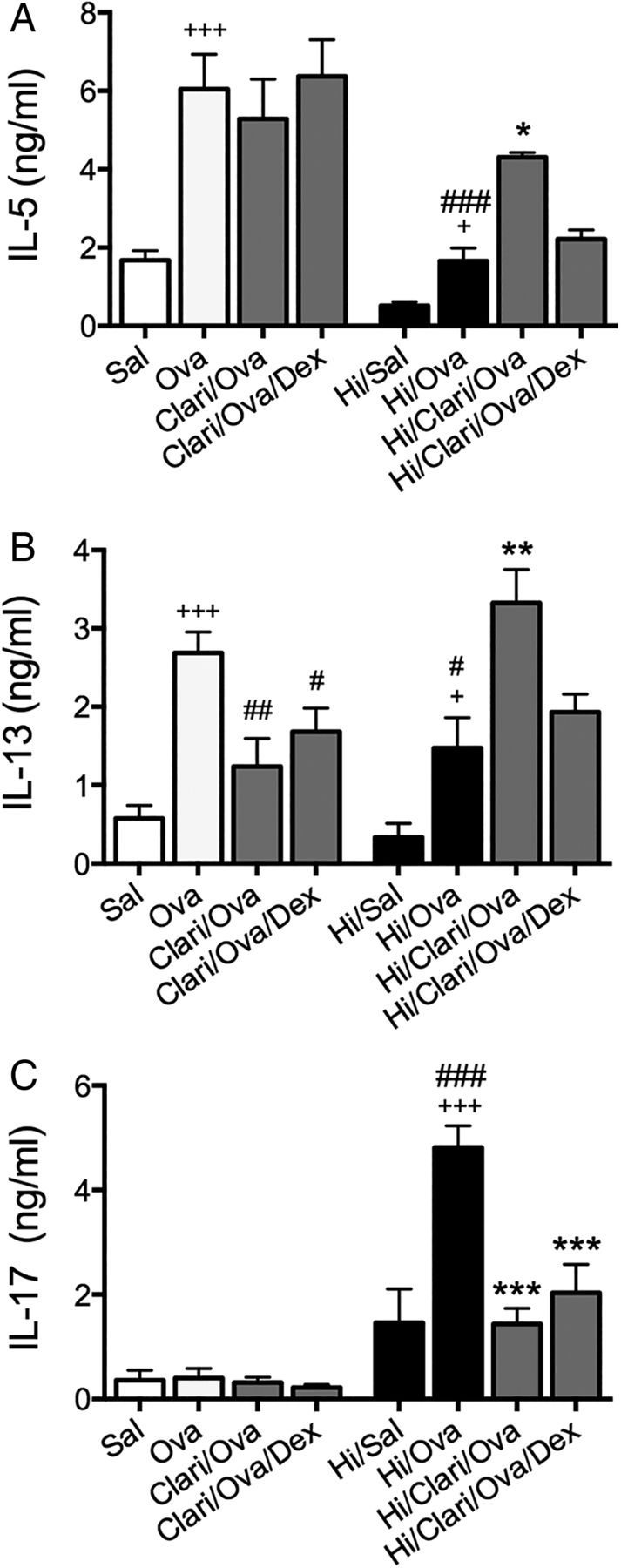
Clarithromycin suppresses interleukin (IL)-17 responses in Haemophilus influenzae (Hi)-induced, severe, steroid-insensitive, neutrophilic allergic airways disease. Effects of clarithromycin treatment on IL-5 (A), IL-13 (B), and IL-17 (C) release from mediastinal lymph nodes. Results are representative of two independent experiments, with a total of six mice/group, and all data are presented as means±SEM. +++p<0.001, +p<0.05 compared with non-allergic (Sal, or Hi/Sal) controls, ###p<0.001, ##p<0.01, #p<0.05 compared with allergic (Ova) controls, ***p<0.001, **p<0.01, *p<0.05 compared with infected, allergic (Hi/Ova) groups.
We next assessed the effect of amoxicillin and clarithromycin treatment on steroid-sensitive AAD and Chlamydia-induced SSIAAD in order to delineate between the antimicrobial and anti-inflammatory effects of macrolide therapy.
Clarithromycin, but not amoxicillin, treatment broadly suppressed the cardinal features of steroid-sensitive AAD
First, we assessed the effects of amoxicillin, an antibiotic with only antimicrobial properties, and clarithromycin, an antibiotic with both antimicrobial and anti-inflammatory properties, on steroid-sensitive AAD (Ova groups).
Amoxicillin treatment (Ova/Amox) did not alter total leucocyte numbers, but reduced neutrophils and eosinophils and, surprisingly, increased macrophage numbers in BALF compared with untreated controls (Ova, figure 2A–D). Treatment had no effect on AHR (airways resistance, figure 2E). Amoxicillin had no additive beneficial effects with DEX treatment (Ova/Amox/Dex), which reduced airway total cells, neutrophils, eosinophils, macrophages and airways resistance.
Clarithromycin treatment alone (Ova/Clari) significantly reduced total cells, neutrophils and eosinophils in BALF as well as AHR (figure 2A–E). Similar reductions also occurred with clarithromycin in combination with DEX (Ova/Clari/Dex).
Clarithromycin, but not amoxicillin, treatment broadly suppressed the cardinal features of Chlamydia-induced SSIAAD
Next, we assessed the effect of amoxicillin and clarithromycin treatments on Chlamydia-induced SSIAAD. We first showed that 3–10 mg/kg doses of both antibiotics had the same effects on infection (see online supplementary figure S1A). Thus, 5 mg/kg of either clarithromycin or amoxicillin was administered on d17–21 of the model (figure 1A).
Amoxicillin treatment alone in SSIAAD (Ova/Cmu/Amox) reduced BALF neutrophils and eosinophils, but had no effect on total cells or macrophages compared with untreated controls (Ova/Cmu, figure 3A–D). Treatment again had no effect on AHR (figure 3E). When amoxicillin was combined with DEX (Ova/Cmu/Amox/Dex), treatment resulted in reductions in all BALF cells and AHR. Since DEX alone did not suppress total cell, neutrophil, eosinophil or macrophage numbers in BALF or AHR in SSIAAD, it is likely that amoxicillin restores sensitivity to DEX.
In contrast to amoxicillin, clarithromycin treatment (Ova/Cmu/Clari) reduced total cells, neutrophils and macrophages in BALF as well as AHR (figure 3A–E). When clarithromycin was combined with DEX (Ova/Cmu/Clari/DEX), treatment suppressed total leucocytes, eosinophils, macrophages and AHR.
Thus, amoxicillin alone is unable to broadly suppress inflammation and AHR but does restore steroid-sensitivity in Chlamydia-induced SSIAAD. Clarithromycin alone broadly suppresses steroid-sensitive AAD and SSIAAD.
Amoxicillin, but not Clarithromycin, inhibits steroid-induced reactivation of infection
We then assessed the effect of antibiotic treatment on the persistence/reactivation of infection, by determination of Cmu 16S expression in whole lung tissue.23 The infection is typically cleared within 20 days in non-allergic mice (ie, before the second Ova challenges on d32).20 16S levels were low/undetectable in amoxicillin and clarithromycin-treated (Ova/Cmu/Amox and Ova/Cmu/Clari) or untreated groups with SSIAAD (Ova/Cmu; figure 3F). Notably, DEX treatment reactivated the infection in SSIAAD (Ova/Cmu/DEX), but this was prevented in groups that were treated in combination with amoxicillin (Ova/Cmu/Amox/DEX). However, reactivation of infection with DEX was not prevented by clarithromycin.
Clarithromycin, but not amoxicillin, broadly suppresses inflammatory mediators in steroid-sensitive AAD and Chlamydia-induced SSIAAD
To investigate the effect of antibiotic treatment on inflammatory mediator responses, the levels of protein (in MLN culture supernatants) and mRNA expression (in lung tissue) of IL-5, IL-13 and TNF-α were assessed. In steroid-sensitive AAD, amoxicillin decreased the protein levels of IL-13, but had no effect on IL-5 (protein or mRNA), IL-13 mRNA or TNF-α (protein or mRNA; figure 4A–C and G–I). In combination with DEX, amoxicillin also decreased IL-5 and TNF-α protein and mRNA. Clarithromycin significantly reduced IL-5, IL-13 and TNF-α protein and IL-13 mRNA. In combination with DEX, the suppressive effects on TNF-α protein and 13 mRNA were removed.
In SSIAAD, amoxicillin decreased IL-13 protein, but increased IL-5 and IL-13 mRNA, and restored the sensitivity of TNF-α mRNA to DEX (figure 4D–F and J–L). Clarithromycin suppressed TNF-α release and mRNA. This also occurred in combination with DEX although IL-13 mRNA was increased.
These results demonstrate that amoxicillin does not broadly reduce inflammatory cytokine responses in steroid-sensitive or SSIAAD. In contrast, clarithromycin alone suppressed these inflammatory cytokine responses in steroid-sensitive AAD and reduced TNF-α responses in SSIAAD. These findings indicate that TNF-α may be an important factor in driving SSIAAD that is suppressed by clarithromycin treatment.
Anti-TNF-α therapy suppresses Chlamydia-induced SSIAAD
To further investigate the role of TNF-α in SSIAAD, the effects of anti-TNF-α antibody treatment were assessed. Treatment reduced total cells, neutrophils, eosinophils and macrophages in BALF as well as AHR in SSIAAD (Ova/Cmu/anti-TNF-α) back to levels in uninfected allergic controls (Sal/Cmu, figure 5A–E). These results show that TNF-α plays a crucial pathogenic role in Chlamydia-induced SSIAAD.
Clarithromycin suppresses Hi-induced SSIAAD
We have previously shown that Hi infection induces SSIAAD driven by Th17 responses.10 To determine whether clarithromycin has widespread applicability for SSI asthma, we assessed its efficacy in our Hi-induced model. Clarithromycin significantly reduced Hi levels at the peak of infection (see online supplementary figure S1B). In Hi-induced SSIAAD (Hi/Ova), clarithromycin treatment reduced neutrophils in BALF as well as AHR, had no effect on total cells, but increased BALF eosinophils compared with untreated controls (figure 6A–E). However, clarithromycin in combination with DEX significantly reduced total cells, neutrophils and eosinophils, had no effect on macrophages, but suppressed AHR.
Clarithromycin suppresses IL-17 responses in Hi-induced SSIAAD
In Hi-induced SSIAAD, clarithromycin increased IL-5 and IL-13, but the addition of DEX reduced the levels of these factors back to those in untreated controls (figure 7A, B). Importantly, clarithromycin alone and in combination with DEX significantly reduced IL-17 levels (figure 7C).
Thus, clarithromycin suppresses SSIAAD and reverses steroid-insensitivity induced by a different infection in a different protocol, by reducing the Th17 responses that drive that phenotype.
Discussion
Twenty per cent of asthmatics have neutrophilic rather than eosinophilic asthma. Neutrophilic asthma is more resistant to anti-inflammatory corticosteroid treatment and is difficult to manage.24 The mechanisms of pathogenesis of this asthma endotype are poorly understood and effective therapies remain to be defined. We previously showed that Chlamydia and Hi infections that are associated with this endotype induce a switch from an eosinophilic, Th2-mediated disease to a neutrophilic, Th1 and/or Th17-mediated phenotype with suppressed Th2 responses in AAD.10 ,18 ,21 Here, we show that these factors are involved in the pathogenesis of steroid-insensitivity and highlight novel therapeutic strategies.
Asthmatics are more susceptible to infection and this may drive SSI asthma. To better model this scenario, we first developed a novel model of Chlamydia-induced SSI asthma. This model involves the induction of a steroid-insensitive phenotype by infection in established AAD. Here, we demonstrate that amoxicillin alone is able to clear infection in SSIAAD, but does not broadly reduce BALF inflammation, T cell cytokine responses or AHR. It does suppress some features, eosinophils and neutrophils are reduced, which may be specifically modulated by amoxicillin in steroid-sensitive AAD. Interestingly, amoxicillin treatment did restore steroid-sensitivity of airway inflammation and AHR in SSIAAD. This is likely to involve the reduction in infection-induced neutrophils, which we have previously shown to be important in the development of severe neutrophilic AAD.21 However, with infection and neutrophils suppressed, there is still underlying Ova-induced AAD and therefore, DEX treatment is still required in order to suppress the remaining features of disease, including leucocytes in BALF and AHR. We also showed that DEX treatment reactivates Chlamydia infection that is inhibited by amoxicillin. In contrast, clarithromycin treatment alone substantially reduced airway inflammation, T cell responses, AHR and infection in two different models of SSIAAD, with similar effects when combined with DEX. This indicates that clarithromycin could be an effective therapy for steroid-sensitive and SSI asthma.
Studies of steroid-insensitive patients on high doses of steroids have shown that they have no reductions in IL-4 and IL-5 expression in BAL cells, and that there is poor suppression of cytokine and chemokine release from peripheral monocytes and alveolar macrophages.25–28 Significantly, our data showing that steroids reactivate an infection suggest that they may be detrimental in infection-induced SSIAAD. We have previously shown that DEX reactivates Hi infection in AAD,10 and extend these studies here by demonstrating that Chlamydia infection is also reactivated. These studies suggest that alternative therapies are required for steroid-insensitive patients. The mechanisms of steroid-induced reactivation of infection have not been investigated. They are likely due to the suppressive effects of DEX on immune/inflammatory responses that are needed to clear infection, thereby creating an environment where bacteria can proliferate.
Macrolides have been proposed as new therapies for SSI asthma as they have both antimicrobial and anti-inflammatory properties and are in widespread clinical use. However, the relative contributions of these properties to efficacy are not known. To date experimental studies of macrolides have been limited to models of mild-to-moderate steroid-sensitive AAD. To delineate the relevant contributions, we compared the effects of amoxicillin, a purely antibacterial agent, with clarithromycin, an antibiotic with additional anti-inflammatory properties in SSIAAD. Although amoxicillin treatment suppressed infection to the same extent as clarithromycin, it only reduced granulocytic inflammation, IL-13 protein, but not other inflammatory mediators and had no effect on AHR in both steroid-sensitive and SSIAAD. Amoxicillin did restore steroid-sensitivity in steroid-insensitive groups. In contrast, clarithromycin treatment alone broadly reduced both inflammation and AHR in both endotypes. Few clinical studies have examined the effect of macrolide treatment in asthma with evidence of infection, however, those that have been performed have had some success. Treatment of adult asthmatics with evidence of Cpn infection with roxithromycin for 6 weeks, significantly improved lung function.29 In asthmatic children with Cpn infection, clarithromycin reduced the duration and risk of subsequent wheezing episodes.30 Other studies have also shown that macrolide treatment reduces inflammation in asthma. Kraft et al31 showed, in asthmatics that were PCR-positive for Cpn, that clarithromycin treatment reduced IL-5 levels. Despite these data, it remains unclear how macrolides exert their beneficial effects.
We investigated likely mechanisms and immune responses involved in the suppression of disease by clarithromycin. In steroid-sensitive groups, clarithromycin reduced both IL-5 and IL-13, which are important in Th2-mediated asthma.24 ,32 Clarithromycin-induced reductions in these cytokines likely suppress the recruitment of eosinophils to the airways, mucus production and AHR. Trials using anti-IL-5 antibodies such as reslizumab, reduced asthma exacerbations and improved FEV1 compared with placebo groups.24 ,32 A recent trial using lebrikizumab, an anti-IL-13 antibody, showed significant reductions in FEV1 after treatment.32 We showed that IL-13 responses promote bacterial infections and associated SSIAAD,18 ,22 ,33 and macrolides may also have beneficial effects in reducing this susceptibility.
In steroid-insensitive groups, clarithromycin treatment, alone and combined with DEX, reduced TNF-α production. To further investigate its role, we inhibited this cytokine during SSIAAD using monoclonal antibodies. Inhibition of TNF-α suppressed neutrophil and macrophage infiltration and AHR, and in the presence of DEX, also reduced eosinophils. This suggests that TNF-α is crucial for the induction of this asthma endotype, and its suppression by clarithromycin reduced key features of disease. Another recent study by Manni et al34 investigated SSIAAD induced by the adoptive transfer of Th17 cells to show that anti-TNF-α treatment reduced lung compliance and airway inflammation, but not tissue inflammation or AHR. In contrast, infection-induced SSIAAD is not solely driven by IL-17. These studies are clinically relevant as TNF-α is a pro-inflammatory cytokine that is implicated in the pathogenesis of severe asthma.32 The administration of anti-TNF-α antibodies to patients with SSI asthma led to improved lung function and AHR, and quality of life.35 However, there have been concerns about its safety.36 We show that TNF-α responses were substantially increased in SSIAAD, which is in agreement with studies that suggest that anti-TNF-α treatment may be most beneficial in SSI asthma.
Together, our studies show that clarithromycin targets Th2 responses in steroid-sensitive AAD, and TNF-α responses in Chlamydia-induced SSIAAD. Thus, this macrolide may have widespread applicability in the treatment of asthma.
This was further confirmed when we investigated the effects of clarithromycin in a different model of infection-induced SSI asthma. We previously showed that Hi infection also induces an SSIAAD phenotype.18 We show in this model that clarithromycin increased Th2 responses, including IL-5, IL-13 and eosinophils. We suggest that this may occur as Hi infection reduces these responses, and when macrolide treatment suppresses infection, the inhibitory effects on these features are removed. Combined treatment with clarithromycin and DEX had the same effects as treatment in the Chlamydia-induced phenotype and significantly reduced neutrophils, eosinophils and AHR. Importantly, we also showed that clarithromycin significantly reduced IL-17 responses in Hi-induced SSIAAD. We have previously shown that this Hi-induced phenotype is mediated by IL-17 responses and that inhibiting this cytokine with monoclonal antibodies reduced key features of disease.10 Thus, clarithromycin may be effective in this phenotype by suppressing IL-17 production. In a similar manner, our clinical studies have shown that clarithromycin treatment reduced airway neutrophil numbers, and sputum IL-8, neutrophil elastase and matrix metalloproteinase-9 levels in SSI asthma.37
Numerous studies have shown that the immunomodulatory activities of macrolides result from their accumulation inside inflammatory cells and suppress ERK1/2 phosphorylation and NF-κB activation that attenuates inflammatory responses in these cells. This is likely how clarithromycin is suppressing SSIAAD in our studies.38
In conclusion, this study demonstrates that clarithromycin has antimicrobial as well as broad immunomodulatory effects and reduces the dominant inflammatory mediators (eg, TNF-α, IL-17) that drive the different endotypes of AAD. In steroid-sensitive groups, clarithromycin suppressed IL-13 to reduce disease features, whereas in a TNF-α or IL-17 environment, it suppressed these factors to reduce disease features. This study, therefore, shows how clarithromycin may be working to suppress key features of infection-induced SSIAAD. Importantly, these studies have identified the efficacy of this treatment on inflammation and infection, and further promote its use as a therapy for infection-induced SSI asthma.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
-
A-TE and JCH contributed equally.
-
Contributors The work presented here was performed in collaboration with all authors. A-TE and JCH: Hypothesis, conception, design, performance and interpretation of experiments, establishment of in vivo models and writing the manuscript. RYK, JWP, ELB and MRS: Acquisition of in vivo data and revision of the manuscript. JRM: Establishment of in vivo models. JLS, PSF and PGG: Conception, revision of the manuscript. PMH: Senior author and responsible for the overall content, hypothesis, conception, design, interpretation of data and revision of the manuscript.
-
Funding This study was supported by funding from the Hunter Medical Research Institute, The National Health and Medical Research Council of Australia (APP1043174), The Asthma Foundation of NSW and the Australian Research Council (DP0559210).
-
Competing interests None.
-
Ethics approval All experiments were approved by the Animal Ethics Committee, University of Newcastle.
-
Provenance and peer review Not commissioned; externally peer reviewed.