Article Text

Abstract
Background People with cystic fibrosis (CF) are managed differently in the USA and UK providing an opportunity to learn from differences in practice patterns.
Objectives To compare cross-sectional demographics, practice patterns and clinical outcomes between US and UK CF patients.
Methods This was a cross-sectional study using 2010 data from patients in the US Cystic Fibrosis Foundation and the UK Cystic Fibrosis patient registries. The a priori outcome measures of interest were lung function and nutritional status. Descriptive statistics and two sample comparisons were performed. Stratification and multivariable linear regression were used to adjust for confounding.
Results The study cohort included 13 777 children and 11 058 adults from the USA and 3968 children and 3965 adults from the UK. In children, mean body mass index centiles were similar. Lung function (FEV1 and FVC% predicted) was significantly higher in US patients ages 6–25 years of age. In a regression model adjusted for only age, FEV1% predicted was on average 3.31% of predicted (95% CI 2.65 to 3.96) higher in the USA compared with the UK. When adjusted for age, age at diagnosis, gender, pancreatic insufficiency and genotype, FEV1% predicted was on average 3.03% of predicted (95% CI 2.37 to 3.69) higher in the USA compared with the UK These differences persisted despite adjustment for possible confounders. Hypertonic saline and dornase alfa were much more commonly prescribed in US children.
Conclusions Children and young adults with CF have better lung function in the USA compared with the UK despite similar nutritional status.
- Cystic Fibrosis
- Paediatric Lung Disaese
- Clinical Epidemiology
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does lung function differ between patients with cystic fibrosis (CF) in the UK and the USA?
What is the bottom line?
-
We have demonstrated important and significant differences in lung function in CF children and young adults in the USA and the UK with children in the USA having better lung function.
Why read on?
-
Our findings suggest that earlier and more aggressive use of chronic pulmonary therapies may be beneficial.
Introduction
A number of advances in the care and outcomes of people with cystic fibrosis (CF) have occurred over the last two decades. This time span has noted dramatic improvements in survival from a median predicted survival of 28 years in 1990 to 38 years in 2010 in the USA.1 ,2 This improvement has been highlighted by Dodge and colleagues noting dramatic improvements in survival by birth cohorts3 and in a recent analysis of the Cystic Fibrosis Foundation (CFF) Patient Registry.4 Improved survival is likely due to the introduction of new therapeutics,5–8 multidisciplinary care, improved nutritional support and the liberal use of antibiotics. The impact of therapies that treat the basic defect,9 ,10 recently approved therapies11–14 and eradication protocols for Pseudomonas15 ,16 are still unknown but likely to be significant.
International comparisons can be extremely informative when comparing how different treatment approaches or medications impact disease progression as in the comparisons of nutritional outcomes and survival between the Boston and Toronto CF care centres,17 which demonstrated the benefits of a high-fat, high-calorie diet. This work was instrumental in unifying the dietary recommendations for CF across the world.18 Other international comparisons conducted until now are more challenging to interpret due to differences in data collection between different nations.19–21
To further our understanding of the role of therapies early in disease and the role of different healthcare systems on outcomes in CF, we compared the CF populations of the USA and the UK. There were three objectives: (1) to compare age-specific demographic characteristics; (2) to compare cross-sectional clinical characteristics and (3) to determine the age-specific differences in lung function and nutritional status between the patient populations.
Methods
Study population
This study is a cross-sectional analysis of two study populations. The study population included all patients enrolled in the CFF Patient Registry and the UK CF Registry in 2010 with clinical data inputted that year into the respective registries and a confirmed diagnosis of CF.22 Both data sets included CF demographics, as well as clinical data (see online supplement for details). Each site involved in the US CFF Patient Registry obtained approval for human subjects participation in research based on local standards and all patients or legally authorised representatives provided informed consent to be included in the registry. National Health Service (NHS) research ethics approval was granted for the UK CF Registry and each patient or legally authorised representative provided written informed consent for data collection and research. Under the terms of the NHS ethics approval, the UK CF Trust steering committee approved this study. One of the major challenges and a unique aspect of this analysis was harmonising the two data registries to address differences in data elements and differences in the seasonality of data entry by country. The data were de-identified and merged to ensure similar seasonality of clinical encounters and recoding of key variables (see the online supplement: data merging and figure E1a, E1b and E2).
Primary outcome measure and predictor of interest
The primary outcome measure was FEV1 per cent of predicted, the single best predictor of mortality in CF.23–25 All per cent predicted values were recalculated for Caucasians in the merged data employing reference equations from Wang and Hankinson26 ,27 with a sensitivity analysis using the global lung function prediction equations.28 We also performed sensitivity analyses restricting the population to those who were homozygotes for the F508del gene and evaluated the differences in FVC% predicted.
Secondary outcomes, confounders and effect modifiers
Weight and height were converted to the metric system. Body mass index (BMI) was calculated using a standard equation (kg/m2). Weight-for-length and BMI centiles were recalculated using US Centers for Disease Control data reference values.29 The use of pancreatic enzymes was deemed synonymous with pancreatic insufficiency. Sputum microbiology results were categorised as negative, positive (≥2 sputum samples positive in 1 year) or intermittent for each CF pathogen (see online supplement). The use of chronic nebulised antibiotics was defined as the use of any one of several inhaled antimicrobial agents. In a post hoc analysis to understand differences in treatments, we employed a modified treatment intensity score (see online supplement), an additive index of the following treatments: hypertonic saline, aerosolised tobramycin, rhDNase, macrolides, aerosolised colistin and other aminoglycosides.30 National-level treatment differences may not be reflected at the individual centre level. Because of this concern, we created centre-level metrics (median and IQR) for both the USA and the UK to perform additional sensitivity analyses. The following covariates were treated as confounders: age at encounter, gender, age at diagnosis, pancreatic sufficiency, genotype, chronic Pseudomonas infection, chronic methicillin-sensitive Staphylococcus aureus (MSSA) infection, Burkholderia cepacia infection and methicillin-resistant Staphylococcus aureus (MRSA) infection. Age and age strata were treated as effect modifiers along with genotype. Age was handled as a categorical variable grouped by 4 age increments (<12 years, 12–17 years, 18–23 years and ≥24 years) for regression models. For comparisons of microbiology and pulmonary therapy, we performed stratified analysis of people under 18 years and those above 18 years.
Statistical analysis
The primary analyses were performed based on an a priori statistical analysis plan. Descriptive analyses of the characteristics of patients according to country (US vs UK) were conducted. Linear regression models with robust SEs were used to model the association of FEV1% adjusted for key covariates. The primary predictor of interest was country (US vs UK). Multivariate models were adjusted for possible confounders by forcing covariates into the model based on known demographic and clinical confounders. Our prespecified multivariable regression models were restricted to data in white patients. We first assessed whether differences in lung function could be explained by differences in age, gender, age at diagnosis, pancreatic insufficiency and cystic fibrosis transmembrane conductance regulator (CFTR) mutation. An interaction term was used for age and country of origin (UK vs US) in the analyses. Stratified analyses and statistical measures of interaction were used to analyse the relationship of country and the following covariates (gender, age and CFTR mutation classification). In multivariate models, we assessed only first-order interactions and tested for significance using the likelihood-ratio test. All of our models assumed statistical independence of every subject within the study population. Since specific centres have been shown to be linked to better outcomes and presumably better care, we repeated analyses with clustering on centre to allow correlation of lung function in subjects within the same centre. Two-tailed α<0.05 was considered statistically significant for all study analyses. Analyses were conducted using Stata V.13 (College Station, Texas, USA).
Results
Demographics
The study cohort included 13 777 children and 11 058 adults from the USA and 3968 children and 3965 adults from the UK. A number of key differences were found when comparing the populations from 2010 (table 1). The median age of the overall population was significantly higher in the UK (17.9 years compared with 16.2 years, difference: 1.2 (95% CI 0.90 to 1.50), p<0.001) with a higher proportion of men (53.1 vs 51.6%, difference: 1.5% (95% CI 0.2% to 2.8%), p=0.02). The median age of diagnosis was earlier in the UK (95% CI 0.3 to 0.4 years, p<0.001). The racial/ethnic distribution also differed in the two populations, with significantly more Asians and fewer black patients in the UK compared with the US CF population, but overall, the vast majority of the CF populations in both countries were white.
Comparisons of demographic characteristics between UK and US data
Pulmonary and nutritional outcomes
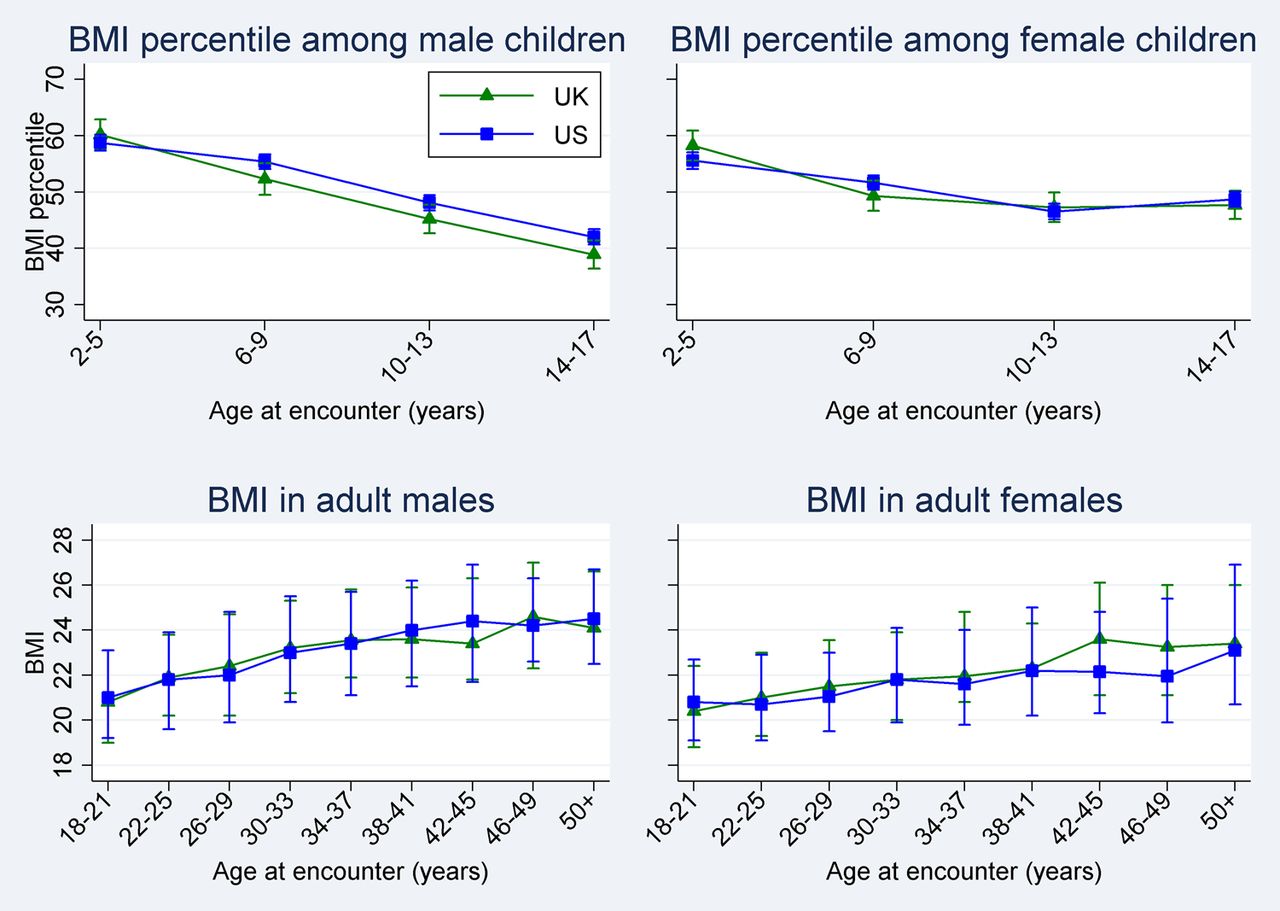
Given the differences noted in the two populations, age-stratified and gender-stratified comparisons were performed for lung function and nutritional status. As noted in Table E1 and figure 1A–D, BMI centile was significantly higher in US male children ages 10–17 years and lung function was significantly higher in US children ages 6–25 years. Interestingly, FEV1% predicted was higher in the UK population that was over the age of 50 years (+5.47% predicted, 95% CI 1.03 to 9.92). Figure 2A clearly denotes the differences between the UK and the USA of FEV1% predicted by age in years (a cross-sectional comparison). The differences in lung function persist when (1) comparing homozygotes for the c.1521_1523delCTT allele in the CFTR gene (F508del) and (2) employing the global lung function prediction equations28 (see online supplement table E2 and figure E3). Additionally, similar patterns were observed when examining FVC% predicted (see figure 2B).

BMI by age and country. Top left: Mean BMI centile (95% CI) among male children aged 2–17 years. Top right: Mean BMI centile (95% CI) among female children aged 2–17 years. Bottom left: Median BMI (IQR) among adult men aged 18 years and older. Bottom right: Median BMI (IQR) among adult women aged 18 years and older.

(A) Mean (95% CI) FEV1% predicted among Caucasian patients by age at clinical encounter and country. (B) Mean (95% CI) FVC % predicted among Caucasian patients by age at clinical encounter and country.
Microbiology
There were modestly higher rates of chronic Pseudomonas aeruginosa infection and markedly lower rates of MRSA and MSSA in the UK compared with the USA (table 2). The higher rate of P. aeruginosa in the UK compared with the USA might be due in part to the age difference. The low rates of MRSA in the UK may be due to differences in antibiotic use in the general population and mirror MRSA rates in other parts of Europe.31
Characteristics of chronic* airway infections in the two countries
Pulmonary therapies
When we evaluated treatments recorded at annual visits for routine care, we found that a number of chronic pulmonary therapies were used much less frequently in the UK compared with the USA (table 3). The most striking differences were noted in the use of hypertonic saline and rhDNase in both children and adults. Chronic macrolide antibiotics were also used less frequently in the UK, but the magnitude of the difference was much less than for hypertonic saline and rhDNase. The overall use of chronic inhaled antibiotics (grouped, given the different antibiotics used in each nation) was similar in the two countries. Because of the stark differences noted in both children and adults, additional analyses were performed stratified by lung function to assess whether these therapies were being preferentially used for those with more advanced disease in the UK. The differences remained in all strata with a general trend of more common use of all medications in both countries in patients with more advanced disease (see online supplement table E3). To capture how medications are used in combination, we employed a modified treatment intensity score.30 In all categories of treatment intensity, mean FEV1% predicted was higher in the USA than in the UK (see online supplement table E4).
Treatment comparisons between the USA and the UK
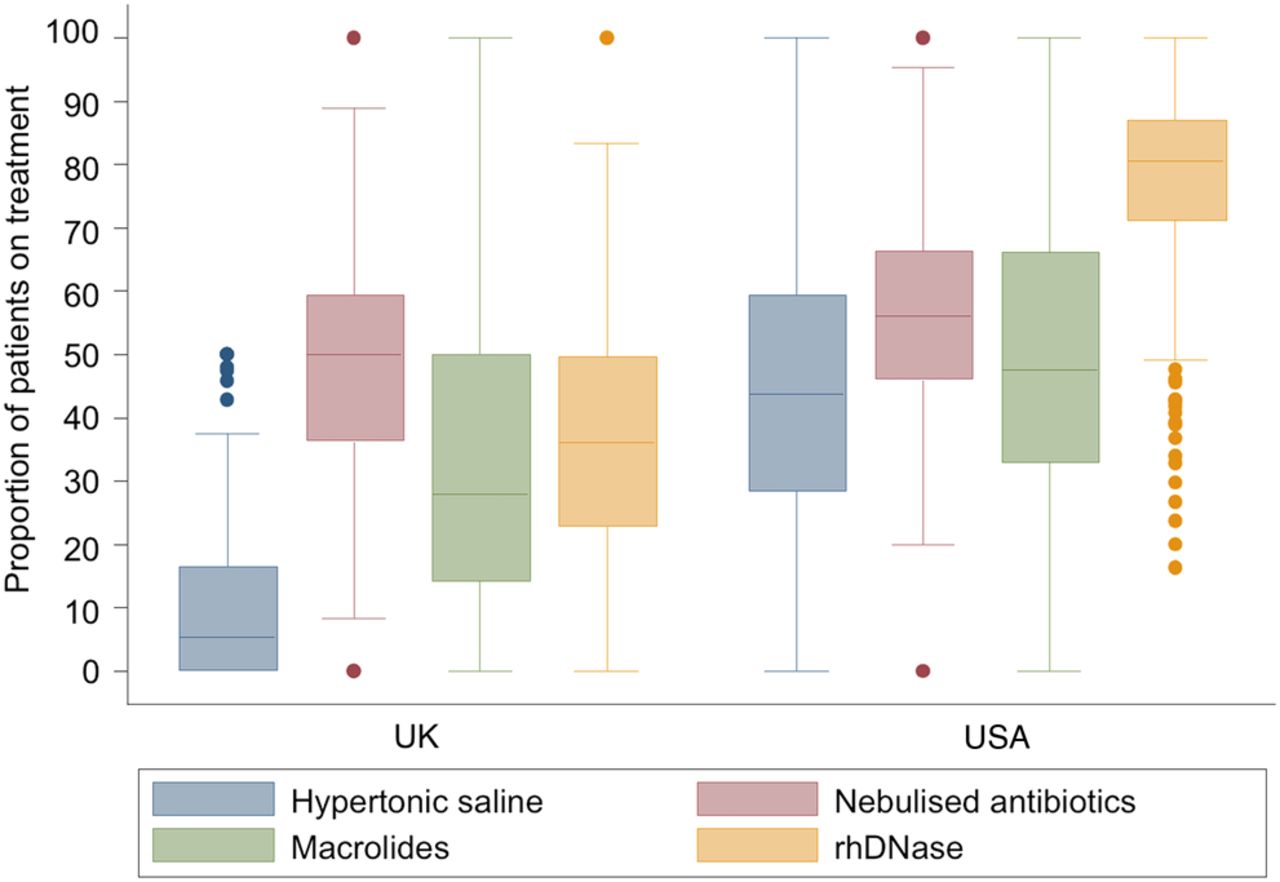
When evaluating our centre-level metrics (median and IQR) for both the USA and the UK, we found that the distribution of treatment rates had a fairly normal distribution in the USA, and that treatment rates at UK centres were significantly skewed to lower treatment rates (figure 3 and online supplement Table E5). This distribution reflects the aggressiveness of care in the USA versus the UK. For children under age 12 years treated with 4 or more therapies, the mean FEV1% predicted in the USA was 85.8% compared with a mean FEV1% predicted of 74.0% in the UK. This suggests that in the USA children with milder lung impairment are treated with more therapies than their counterparts in the UK. Treatments were much less commonly used at a large number of UK CF centres. However, the distribution of use of inhaled antibiotics appeared to be much more similar between the two countries.

{kind=link}
{kind=link}
{kind=link}
Distribution of centre-level treatment rates by treatment and country.
Multivariable statistical models
In a regression model adjusted for only age at encounter, FEV1% predicted was on average 3.31% of predicted (95% CI 2.65 to 3.96) higher in the USA compared with the UK (Model 1) (table 4). When we adjusted for the impact of age, age at diagnosis, gender, pancreatic insufficiency (based on pancreatic enzyme use) and genotype, the FEV1% predicted was on average 3.03% of predicted (95% CI 2.37 to 3.69) higher in the USA compared with the UK (Model 2). Because sputum microbiology could relate to the local environment, we created an additional regression model adjusting for the above-noted variables and sputum microbiology. The results showed a 3.85% predicted (95% CI 3.17 to 4.53) difference in FEV1% predicted between countries (Model 3). In each of these regression models, the effects of the other adjusted covariates mirrored effects seen in other studies.32 ,33 In a post hoc analysis, we employed an interaction term of age versus country; this analysis was driven by prior analyses showing that the majority of the effect between countries was in children. As there was statistically significant evidence of interaction, we ran Model 3 stratified by age group and noted that the difference between UK and US patients was statistically significant only in those under 24 years (table 4). We also conducted a number of analyses first restricting models to only those who were homozygous for the F508del mutation. Differences persisted in these analyses. We also reran the regression models employing raw FEV1 adjusting for height, age and gender and demonstrated clinically significant differences between the US and the UK in lung function (see online supplement table E6 and E7).
Linear regression models of FEV1% predicted among white patients
An additional set of sensitivity analyses were performed to allow correlation of lung function in subjects within the same centre. These models did not differ significantly from any of the earlier models.
Discussion
In a cross-sectional analysis of data from two National CF registries, we have demonstrated stark differences in lung function in children with CF in two countries with well-developed yet different healthcare systems. Lung function as measured by FEV1% predicted was higher in children in the USA compared with those in the UK, and these differences persisted up to the early 20s. The differences were not associated with accompanying differences in nutritional status and persisted in both stratified analyses and in multiple variable adjusted regression models. The most striking differences between the two populations were the low rates of MRSA and MSSA infections in the UK in children and adults and the modestly higher rate of Pseudomonas infection in children in the UK. The low rates of MSSA noted in the UK could be due to the common practice of Staphylococcus prophylaxis used in the first 3 years of life. The results noting higher MRSA in the USA but higher lung function appear to contradict earlier analyses regarding the role of MRSA on survival and lung function by Dasenbrook and colleagues noting worse lung function and higher mortality in those with persistent MRSA.34 ,35 However, Sawicki and colleagues found the opposite results—that incident MRSA had no impact on lung function decline but was a marker of more intensive treatement.36 The differences noted between the two countries were not due to different or over-representation of milder mutations, given the concordant findings in those analyses restricted to those patients who were homozygous for the F508del mutation. The differences were also not due to differences in age distribution of the populations or age of diagnosis. The striking difference in practice patterns between the two populations was in the rate of use of several pulmonary therapies. US centres had on average a higher intensity of therapy compared with the UK.
Comparing clinical outcomes between countries can be very informative, particularly where clear differences in care models and treatment approaches occur. Survival differences and their association with nutritional approach noted between Toronto and Boston led to a complete revision of the nutritional model of care throughout the world.17 Recent analyses of the European CF Registry noted marked differences in the median age in the European Union (EU) compared with non-EU countries with far fewer CF patients over age 40 living in non-EU countries.20 One recent analysis that addressed treatment differences focused on differences between the USA and Australia.19 This analysis noted the benefit of newborn screening on lung function was significantly less in Australian children compared with US children, and mean FEV1% predicted adjusted for age, gender and genotype did not differ between the two countries. These studies have demonstrated the potential benefit of international comparisons of clinical outcomes in CF.
A number of national comparisons have been done primarily to compare survival.21 ,37 Much of this work has focused on issues related to comparing survival metrics between countries and addressing some of those challenges. While these initial demographic and survival comparisons are interesting, to improve our understanding of how to best manage CF patients, we need to assess more proximal outcomes like lung function and nutrition with accompanying analysis of treatments. Differences in treatment strategies and treatment approaches may lead to changes in intermediate outcomes such as lung function and nutrition. Our analysis has clearly demonstrated significant lung function differences between the two countries seen primarily in children. Although the two patient registries use similar software for data entry, the countries differed in frequency of data collection. We paid special attention to this key difference by using random sampling methodology in the US data to match seasonal differences in data entry. Earlier comparisons have not addressed this important potential confounder.19 A number of earlier studies have demonstrated the role of seasons on exacerbation frequency in relation to seasonal variation in respiratory viruses and in relation to pseudomonal acquisition.38–40 Our analysis is the first to carefully ensure that differences in timing of clinical assessment do not confound potential associations.
Our results demonstrate striking differences in the use of CF therapies in the two countries, particularly in children. In the UK, universal access to care is available, while in the USA that is not the case. Because of universal access to care, the UK employs a reimbursement system for CF specialised care based on tariffs linked to disease severity41 in addition to careful review of the cost-effectiveness of medications via the UK's National Institute of Health and Care Excellence (NICE).42 In the USA, medications are much more likely to be used outside of the confines of the study populations defined in the pivotal clinical trials in CF leading to dramatic increases in cost between 2001 and 2007.43 While such use of therapeutics is emblematic of US healthcare and likely has not yielded improved outcomes,44 in the case of CF, this intensity of use of therapeutics may confer benefit, particularly in children with the disease.45 While use of therapeutics was more common in adults in the USA compared with the UK, these differences did not translate into improved lung function. One could argue that prescribing patterns in the UK are more efficient to achieve similar lung function with fewer therapies. This pattern, however, was not seen in children.
Our analyses have a number of limitations. The first limitation is the fact that this analysis is a cross-sectional analysis, a weaker study design that limits causal inference. Both temporal changes and cohort effects can conflate the results, particularly if sicker subjects are dying leaving a healthier population. Give the very low death rate in children with CF, this is unlikely to account for our findings. This potential weakness, however, does not diminish the significance of our findings. Our results point to stark differences in lung function over many years of age, and these differences are coupled with very different treatment patterns. The direction of the bias for treatment intensity should have been opposite of our findings—the USA should have had lower lung function if treatment was directed at more severe patients. We found the reverse association. An additional limitation that deserves mention is neither registry is likely to capture every CF patient residing in each nation. If nations have differential sampling of the CF population, our results may merely reflect that differential sampling. Prior data, however, support the finding that those not captured in the US CFF Patient Registry are more likely to be post lung transplant, thus not impacting the findings of this analysis.46 ,47 An additional potential bias could be due to differential capture of atypical or mild CF in the USA compared with the UK. This is extremely unlikely given that the subgroup analysis in patients who were homozygous for the F508del mutation replicated the main analysis. Of note, data related to adherence to therapies are not available. Thus, when patients are noted in the respective registries of being on a therapy, we do not know if they merely trialled the therapy and then stopped it. This limitation cannot be overcome in the current registry data; however, this potential misclassification is unlikely to be differential. Lastly, the differences that we found could be due to differences in socioeconomic status.48 ,49 Unfortunately, measures are not available that can easily compare the socioeconomic status of those CF patients living in the USA and the UK.
Conclusion
We have clearly demonstrated stark differences in lung function as measured by FEV1% predicted between the USA and the UK in children and young CF adults. These differences in lung function persisted with a number of sensitivity analyses and in multivariable adjustment for confounders. The differences were associated with very significant differences in the aggressiveness of care, particularly in CF children, which may have long-term implications to outcome in this disease. Further longitudinal comparisons of national data are needed to unravel the causal implications of earlier and more aggressive treatment of CF children.
Acknowledgments
We would like to acknowledge the support of the US CFF and the UK CF Trust that made this analysis possible. We would also like to acknowledge all the CF patients in both the USA and the UK who consent to be part of their respective National Patient Registries. Last, we would like to acknowledge Ase Sewall of Sewall, Inc., Bethesda, for her work on the US CFF Patient Registry.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CHG, SJM, HBQ, BCM, AE, EAK, KP, EG, JO and DB contributed to the conception and design of the study. AE merged the study data. CHG, SJM, HBQ, BCM, AE, EAK, KP, EG, JO and DB contributed to the analysis and interpretation of the data. CHG drafted the article. CHG, SJM, HBQ, BCM, AE, EAK, KP, EG, JO and DB revised the article critically for important intellectual content. All authors contributed to the final version of the article and approval of the final version to be published.
-
Funding This analysis was supported with funds from the UK CF Trust and the US CF Foundation. CHG receives funding from the US NIH, FDA and the European Commission (R01HL103965, R01HL113382, R01AI101307, U M1HL119073 and P30DK089507) and the FDA (R01FD003704). DB is supported by NIHR funding to the Respiratory Biomedical Research Unit at Royal Brompton Hospital and Imperial College London, UK.
-
Competing interests CHG has received grant funds from the US CFF. He has also received funds from F. Hoffmann-La Roche Ltd for an honorarium for preparation and giving a symposium talk, an unrestricted grant from Vertex Pharmaceuticals to conduct secondary data analysis, an honorarium from Gilead Sciences to participate in the Gilead Scientific Scholars Grant Review Panel, an honorarium from Johns Hopkins University for continuing education materials and a talk. He has received funding from the US NIH, FDA and the European Commission for cystic fibrosis research. CHS received funding from the CFF, the NIH (R01HL103965, R01HL113382, R01AI101307, U M1HL119073 and P30DK089507) and the FDA (R01FD003704). DB is supported by NIHR funding to the Respiratory Biomedical Research Unit at Royal Brompton Hospital and Imperial College London, UK. US CFF Patient Registry is funded by the US CFF. UK CF Registry is funded by the UK CF Trust.
-
Ethics approval Each site involved in the US CFF Patient Registry obtained approval for human subjects participation in research based on local standards and all patients or legally authorised representatives provided informed consent to be included in the registry. NHS research ethics approval was granted for the UK CF Registry and each patient or legally authorised representative provided written informed consent for data collection and research. Under the terms of the NHS ethics approval, the UK CF Trust steering committee approved this study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The data published in this paper are held by both the US CFF and the UK CF Trust. These data are not publicly available, but permission for data analyses can be obtained by successful application to each of the respective organisation. Both organisations use peer review of scientific proposals prior granting access to anonymised data sets.