Article Text

Abstract
Background We tested the hypotheses that fibrinogen and α1-antitrypsin are observationally and genetically associated with exacerbations in COPD.
Methods We studied 13 591 individuals with COPD from the Copenhagen General Population Study (2003–2013), of whom 6857 were genotyped for FGB -455 (rs1800790, G>A) and FGB -448 (rs4220, G>A) and had plasma fibrinogen measured. Furthermore, 13 405 individuals were genotyped for the SERPINA1 S-allele (rs17580) and the Z-allele (rs28929474) and had measurements of plasma α1-antitrypsin. Exacerbations were defined as hospital admissions or treatments with systemic corticosteroids. We studied observational associations between plasma measurements and exacerbations in Cox regression analyses, associations between genotypes and exacerbations in logistic regression analyses and associations between genetically determined plasma levels and exacerbations in instrumental variable analyses.
Results Elevated fibrinogen and α1-antitrypsin levels were associated with increased risk of exacerbations in COPD, HR=1.14 (1.07 to 1.22, p<0.001) and 1.18 (1.11 to 1.25, p<0.001), respectively, per SD increase. Presence of the Z-allele was associated with increased odds of exacerbations, OR=1.25 (1.05 to 1.48, p=0.01), as was α1-antitrypsin level genetically lowered by the Z-allele, OR=1.07 (1.02 to 1.13, p=0.004), per SD decrease. Fibrinogen elevating genotypes, FGB -455 (AA) and FGB -448 (AA), were not associated with exacerbations, OR=0.96 (0.73 to 1.25, p=0.77) and OR=1.01 (0.75 to 1.33, p=0.90), respectively, and neither was genetically elevated fibrinogen level, OR=1.11 (0.76 to 1.63, p=0.58) per SD increase.
Conclusions Fibrinogen and α1-antitrypsin were observationally associated with increased risk of exacerbations. However, genetically, fibrinogen per se was not associated with exacerbations, while lowered α1-antitrypsin was associated with increased odds of exacerbations.
- COPD epidemiology
- COPD Exacerbations
Statistics from Altmetric.com
Key messages
What is the key question?
Are elevated levels of plasma fibrinogen and plasma α1-antitrypsin both observationally and genetically associated with exacerbations of COPD?
What is the bottom line?
Plasma fibrinogen and plasma α1-antitrypsin are positive acute phase proteins and, observationally, elevated levels are likely associated with an increased risk of exacerbations in COPD.
Why read on?
We confirmed that observationally elevated levels of the acute phase proteins fibrinogen and α1-antitrypsin are associated with an increased risk of exacerbations in COPD, but in contrast to observational findings we observed that it was genetically lowered α1-antitrypsin that was associated with an increased risk of exacerbations, while genetically elevated fibrinogen was not, indicating that fibrinogen per se is not causally associated with exacerbations while α1-antitrypsin is.
Introduction
COPD, which is now the third leading cause of death in the world,1 is characterised by airflow limitation, breathlessness and exacerbations.1 ,2 Exacerbations are important events with a significant influence on prognosis, and prevention of exacerbations is a central element in the management of COPD.3–6
Systemic inflammation and elevated inflammatory biomarkers have been linked to increased risk of exacerbations in COPD,4 and elevated plasma fibrinogen is an example of a strong predictor of increased risk of exacerbations.7 Therefore, plasma fibrinogen is gaining increased interest and is currently considered for approval by the US Food and Drug Administration (FDA) as a way to stratify individuals with COPD in clinical trials,8 through efforts by the COPD Biomarker Qualification Consortium.9 However, it is not known whether both observationally and genetically elevated plasma fibrinogen are associated with exacerbations of COPD. These possible differences can be assessed using a Mendelian randomisation design studying associations between genotypes, genetically elevated or lowered plasma measurements and exacerbations. As an example of discrepancy between observational and genetic associations, previous studies have shown that observationally elevated plasma C reactive protein (CRP) is a strong predictor of increased risk of COPD,7 ,10 whereas genetically elevated levels of plasma CRP are not associated with COPD.11 This suggests that elevated plasma CRP per se is not causally associated with COPD.
Plasma α1-antitrypsin is an acute phase protein,12 and elevated levels of this inflammatory biomarker will on a population level likely be associated with increased risk of exacerbations in COPD. However, from previous studies we know that it is the genetically lowered levels of plasma α1-antitrypsin that are associated with increased risk of emphysema.13 ,14 It is not known whether genetically lowered plasma α1-antitrypsin levels are also associated with exacerbations in COPD in the general population.
We tested the hypotheses that fibrinogen and α1-antitrypsin are both observationally and genetically associated with exacerbations in COPD. For comparison we included plasma CRP as an observational but not genetic predictor of exacerbations.
Methods
Study population
The Copenhagen General Population Study is an ongoing population-based study initiated in 2003 to study individuals from the suburbs of Copenhagen.10 ,15 We used data on 96 603 individuals examined from 2003 to 2013 with pulmonary function tests and a blood sample drawn at the day of examination (see e-figure 1). Of these, we identified 13 591 individuals with COPD (airflow limitation defined by FEV1/FVC<0.7, no asthma and age above 40 years). From 2003 to 2009, a total of 6857 individuals with COPD were genotyped for FGB -455 and FGB -448 fibrinogen genotypes and had plasma fibrinogen measured. Furthermore, from 2003 to 2011 a total of 10 268 individuals with COPD had CRP genotype determined and plasma CRP level measured. Finally, from 2003 to 2013 a total of 13 405 individuals with COPD had α1-antitrypsin genotypes determined and plasma α1-antitrypsin measured. The different numbers of consecutive individuals genotyped for the different genetic variants only reflect that genotyping was conducted in 2009, 2011 and 2013, respectively, for the number of individuals recruited to the Copenhagen General Population Study at that time.
Genotypes
We used single nucleotide polymorphisms (SNPs) in the β-fibrinogen promoter FGB -455 (rs1800790, G>A) and exon FGB -448 (rs4220, G>A).16 ,17
As mentioned, CRP was included as a control in our study. The SNP in the CRP promoter (rs3091244, C>T and/or C>A) was a priori expected to be strongly associated with elevated plasma CRP levels, but not with exacerbations.11 ,18–20 Due to the low number of individuals with the AA genotype (N=19), we pooled the AA genotype with the AT genotype.
For α1-antitrypsin we studied the S-allele and Z-allele exon SNPs in the SERPINA1 gene (rs17580 and rs28929474, M>S and M>Z).21 Due to the low number of individuals with COPD and the α1-antitrypsin genotypes SS (N=15), SZ (N=15) and ZZ (N=17), we pooled genotypes, studying the MM genotype as reference group versus having at least one S-allele but no Z-allele (MS and SS) and those having at least one Z allele (MZ, SZ and ZZ) together.
Genotypes are expected to be largely unconfounded,22 since genotypes are randomised at conception. The α1-antitrypsin Z genotype deviated from Hardy-Weinberg equilibrium (p=0.005), but all other genotypes were in Hardy-Weinberg equilibrium (p=0.35 for FGB-455, p=0.66 for FGB-448, p=0.60 for rs3091244 and p=0.52 for the S-allele; χ2 tests).
Plasma markers
Plasma levels of fibrinogen (in μmol/L), high-sensitivity CRP (in mg/L) and α1-antitrypsin (in μmol/L) were measured in blood samples drawn at the examination date in the Copenhagen General Population Study. Fibrinogen was measured in fresh samples by a turbidimetric prothrombin time-derived method.16 High-sensitivity CRP was measured on fresh samples using turbidimetry or nephelometry.23 α1-Antitrypsin was measured by an immunoturbidimetric assay (Konelab, Helsinki, Finland).
Exacerbations
Exacerbations in COPD were defined by a composite of hospital admissions with a discharge diagnosis of COPD in the national Danish Patient Registry24 and/or dispensed treatments with systemic corticosteroids alone or in combination with antibiotics in the national Danish Medicinal Product Registry.5 ,25 Linkage between the Copenhagen General Population Study and these nationwide registers were done using the unique personal civil registration system in Denmark.26 In the national Danish Patient Registry we had access to data from the initiation of the registry in 1977, and in the national Danish Medicinal Product Registry we had access to data from the initiation in 1995. Since genotype exposure has been present in each individual since conception, we studied exacerbations during full follow-up until end of data access in 2013 (end of follow-up) in our genetic analyses. All individuals were followed up during the entire period, and death (N=1189; 8.7%) or emigration (N=16; 0.1%) after the examination date among individuals with COPD led to censoring at the specific date.
Statistical analyses
Demographic analyses, linear regression analyses, logistic regression analyses and Cox regression analysis were used from the statistical software package R (V.3.0.2).27 ,28 Instrumental variable analyses were done using the ‘ivprobit’ function in the statistical software package Stata (V.13).29
Figure 1 shows the study design.

Study design.
Observed plasma levels and exacerbations
We studied observational associations between elevated plasma fibrinogen and elevated plasma α1-antitrypsin and time to first exacerbation during a maximum of 3 years from the baseline examination using Cox regression analyses.30 We included age at examination, sex, smoking (current/former/never), breathlessness (a score on the modified Medical Research Council scale, mMCR≥2) and FEV1% of predicted as confounders.
Genotypes and plasma levels
To analyse the robustness of the associations between genotypes and the levels of corresponding plasma markers we used univariable linear regression analyses. Since plasma CRP levels are highly skewed, we used log(CRP) as outcome in linear regression analyses. Therefore, we report results both as geometric means and as the arithmetic mean of plasma levels of CRP. To provide an easy visual interpretation of relative differences in plasma levels caused by the different genotypes, we also calculated the effects of genotypes on differences in plasma levels in SDs.
Genotypes and exacerbations
To analyse possible associations between genotypes and odds of having at least one exacerbation during full follow-up we applied logistic regression analyses.30
Genetically determined plasma levels and exacerbations
To estimate associations between genetically elevated plasma fibrinogen and genetically lowered plasma α1-antitrypsin we used a two-stage instrumental variable regression model. The basics of this Mendelian randomisation design are described in figure 1.22 ,31–33 In these analyses, we used either fibrinogen genotypes FGB-455 and FGB-448, or the Z-allele and the S-allele as our genetic instruments and elevated fibrinogen or lowered α1-antitrypsin levels, respectively, as our endogenous regressors. We applied a two-stage probit model, using the ‘ivprobit’ function in the statistical software package Stata.29 ,34 To test for endogeneity we used the Durbin–Wu–Hausman test for the hypothesis that the two-stage probit model was necessary,35 and to test for the strength of instruments we used the F-tests from the first-stage regressions (see e-table 1 in the online supplementary data).36 ,37
Study approval
The study was approved by Herlev Hospital and a Danish regional ethics committee (H-KF01-144/01) and was conducted according to the Declaration of Helsinki. Written informed consent was obtained from all individuals.
Results
Demographics
Plasma fibrinogen levels in COPD were elevated compared with plasma fibrinogen levels in individuals without COPD from the Copenhagen General Population Study, 12.2 μmol/L (SD=3.1) versus 11.3 μmol/L (SD=2.8), (p<0.001; one-way analysis of variance (ANOVA)). Furthermore, plasma α1-antitrypsin levels in COPD were also elevated compared with individuals without COPD from the Copenhagen General Population Study, 24.2 μmol/L (SD=4.6) versus 23.3 (SD=4.5), (p<0.001), reflecting the positive acute phase protein attribute.
Table 1 and e-tables 2–7 in the online supplementary data show characteristics of the 13 591 individuals with COPD in the Copenhagen General Population Study. Table 1 also shows statistical associations with either genotypes or the observational plasma marker tertiles, with details provided in e-tables 2–7 in the online supplementary data. None of the variables were associated with the fibrinogen genotypes FGB -455 (G>A) and FGB -448 (G>A), whereas most variables were observationally strongly associated with plasma fibrinogen tertiles. Likewise, in our control analyses, CRP genotype was not associated with any variables, but observationally the tertiles of plasma CRP showed strong associations with most variables. For α1-antitrypsin genotype we observed an expected strong association with FEV1 (p<0.001), but no confounding by other variables. In addition, we found that all variables were strongly observationally associated with plasma α1-antitrypsin tertiles, reflecting the positive acute phase reactant attribute of α1-antitrypsin.
Characteristics of participants in the Copenhagen General Population Study (2003–2013) among a total of 13 591 individuals with COPD
E-tables 8–14 in the online supplementary data show characteristics for all variables among all 96 603 individuals in the Copenhagen General Population Study cohort and, additionally, all associations between variables, genotypes and observational plasma tertiles.
Observed plasma levels and exacerbations
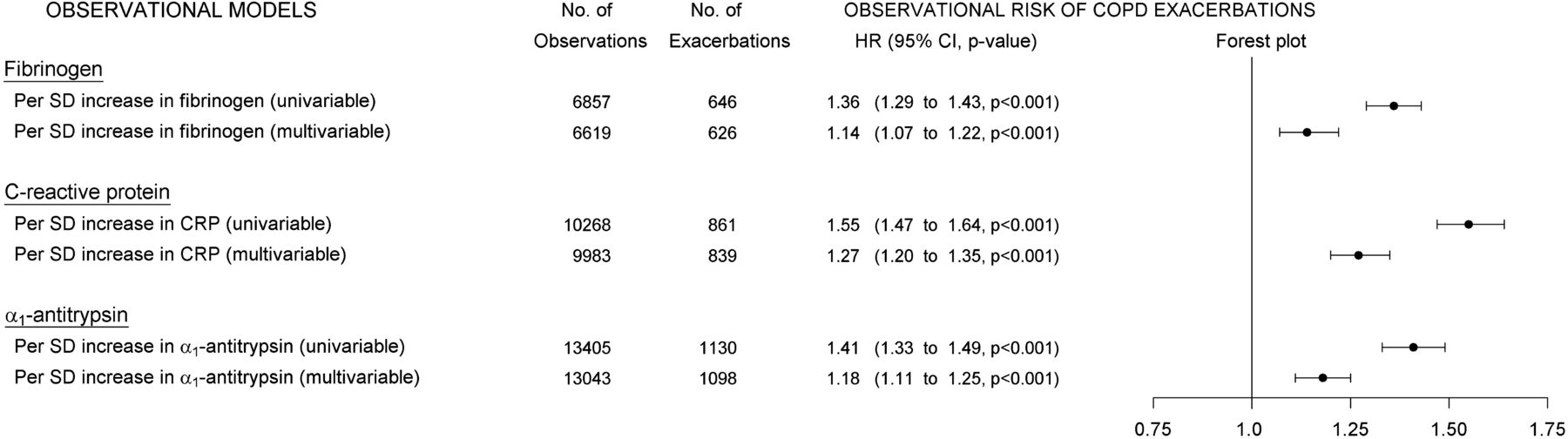
Observational Cox regression analysis confirmed that elevated plasma fibrinogen was associated with an increased risk of exacerbations, HR=1.14 (95% CI 1.07 to 1.22, p<0.001), per SD increase in plasma fibrinogen (upper part of figure 2).

Observational plasma levels and exacerbations in COPD. Cox regression analyses showing associations between elevated plasma levels and risk of exacerbations in COPD. From left to right: univariable or multivariable models for each inflammatory biomarker (‘OBSERVATIONAL MODELS’), number of individuals in corresponding analyses (‘No. of Observations’), number of exacerbations during follow-up (‘No. of Exacerbations’), hazards ratios with 95% CIs and p values (‘HR (95% CI, p value)’) and a corresponding forest plot (‘Forest plot’).
As expected, observationally elevated CRP was also strongly associated with increased risk of exacerbations (middle part of figure 2).
Furthermore, analysing plasma α1-antitrypsin as a continuous variable on a population level we observed that observationally elevated plasma α1-antitrypsin was associated with an increased risk of exacerbations in COPD, HR=1.18 (1.11 to 1.25, p<0.001), per SD increase in plasma α1-antitrypsin (lower part of figure 2).
Genotypes and plasma levels
In linear regression analyses among individuals with COPD, FGB -455 (AA) and FGB -448 (AA) homozygosity was robustly associated with increased levels of plasma fibrinogen, +0.87 μmol/L (95% CI 0.50 to 1.23, p<0.001) and +0.94 μmol/L (0.54 to 1.34, p<0.001), respectively, compared with the GG genotype (upper part of figure 3).

Genotypes and plasma levels in COPD. Robustness of associations from linear regression analyses between genotypes and plasma levels in COPD. From left to right: genotypes (‘Genotype’), number of individuals in study (‘No. of Observations’), β-coefficients from linear regression analyses with 95% CI (‘Beta 95% CI’), differences in plasma levels calculated in SDs (‘SD’), p values from test-for-trend (‘p<0.001’) and arithmetic mean plasma levels (‘Plasma means’). Please note that the ‘Plasma means’ for C-reactive protein (CRP) in the figure are reported on the original measurement scale as arithmetic means and not as geometric means from the regression analyses.
As expected, the CRP genotype was robustly associated with differences in plasma CRP levels, with the AT/AA genotype most strongly associated with elevated plasma CRP, +1.31 mg/L (1.18 to 1.45, p<0.001, geometric mean), compared with the CC genotype (middle part of figure 3).
For α1-antitrypsin genotype, we observed that having a least one Z-allele was robustly associated with lowered levels of plasma α1-antitrypsin, −9.52 μmol/L (−9.22 to −9.82, p<0.001) compared with the MM genotype. Furthermore, having at least one S-allele but no Z-allele was also robustly associated with lowered levels of plasma α1-antitrypsin, −3.76 μmol/L (−3.46 to −4.06, p<0.001) compared with the MM genotype (lower part of figure 3).
Genotypes and exacerbations
Homozygosity for the plasma fibrinogen elevating genotypes FGB -455 (AA) and FGB -448 (AA) was not associated with exacerbations in COPD in logistic regression analyses, OR=0.96 (0.73 to 1.25, p=0.77), and OR=1.01 (0.75 to 1.33, p=0.90), respectively, compared with the GG genotype (upper part of figure 4).

Genotypes and odds of exacerbations in COPD. Associations from logistic regression analyses between genotypes and odds of exacerbations in COPD. From left to right: genotypes (‘Genotype’), number of individuals in study (‘No. of Observations’), number of individuals with exacerbations (‘No. of Exacerbations’), OR from logistic regression analyses with 95% CI and p value (‘OR (95% CI, p value)’) and a corresponding Forest plot.
As expected, the plasma CRP-elevating genotype was not associated with exacerbations in COPD (middle part of figure 4).
In contrast, having at least one plasma α1-antitrypsin-lowering Z-allele (MZ, SZ or ZZ) was associated with increased odds of exacerbations in COPD, OR=1.25 (1.05 to 1.48, p=0.01) compared with the MM genotype (lower part of figure 4). However, presence of the α1-antitrypsin-lowering S-allele (MS or SS) was not associated with exacerbations, OR=0.96 (0.80 to 1.14, p=0.64) compared with the MM genotype. Adjustment for FEV1 only changed our estimates slightly, OR=1.23 (1.03 to 1.47, p=0.02) and OR=0.98 (0.81 to 1.17, p=0.81), for the Z-allele and the S-allele, respectively.
Genetically determined plasma levels and exacerbations
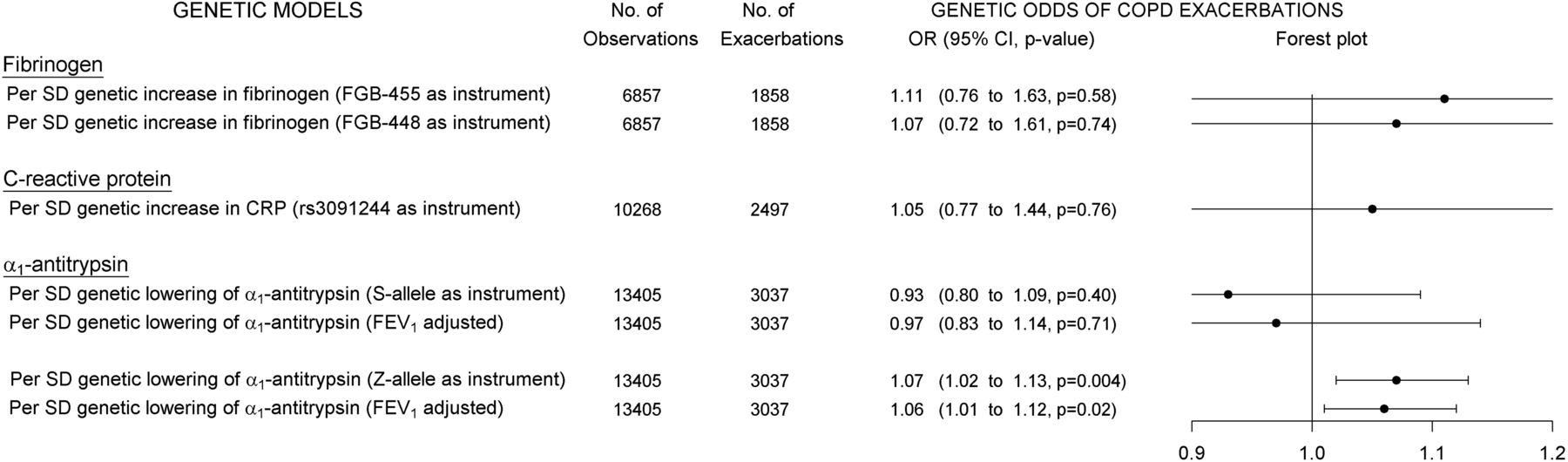
Instrumental variable analyses showed that genetically elevated levels of plasma fibrinogen were not associated with exacerbations, OR=1.11 (0.76 to 1.63, p=0.58) and OR=1.07 (0.72 to 1.61, p=0.74) per SD increase in plasma fibrinogen using FGB-455 or FGB-448, respectively, as instruments (upper part of figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genetic plasma levels and exacerbations in COPD. Instrumental variable analyses showing associations between genetic differences in plasma levels and odds of exacerbations in COPD. From left to right: Instrumental variable models for each genotype and corresponding inflammatory plasma biomarker (‘GENETIC MODELS’), number of individuals in corresponding analyses (‘No. of Observations’), number with exacerbations during full follow-up (‘No. with Exacerbations’), ORs with 95% CIs and p values (‘OR (95% CI, p value)’) and a corresponding forest plot (‘Forest plot’).
As expected, genetically elevated CRP was not associated with exacerbations (middle part of figure 5).
In contrast, further instrumental variable analyses estimated that plasma α1-antitrypsin genetically lowered by the Z-allele was significantly associated with increased odds of exacerbations, OR=1.07 (1.02 to 1.13, p=0.004) per SD decrease in plasma α1-antitrypsin (lower part of figure 5). Adjustment for FEV1 only changed this estimate slightly, OR=1.06 (1.01 to 1.12, p=0.02). Plasma α1-antitrypsin genetically lowered by the S-allele was not associated with exacerbations, OR=0.97 (0.83 to 1.14, p=0.71) per SD decrease in plasma α1-antitrypsin (lower part of figure 5).
A summary of all fibrinogen and α1-antitrypsin results are shown in e-figures 2 and 3.
Discussion
In this study of 13 591 individuals with COPD, we observed that elevated plasma fibrinogen and elevated plasma α1-antitrypsin were associated with increased risk of exacerbations, reflecting their mutual positive acute phase protein attribute. In contrast, genetically lowered plasma α1-antitrypsin was associated with increased odds of exacerbations, while genetically elevated plasma fibrinogen was not associated with exacerbations. These findings are novel.
Numerous studies have shown that elevated plasma fibrinogen is a strong predictor of exacerbations in COPD.10 In this study, we confirmed these observations. However, we bring suggestive evidence that elevated plasma fibrinogen per se is not causally associated with exacerbations in COPD, since genetically elevated plasma fibrinogen was not associated with exacerbations at all. Although this finding is novel, it may not come as a surprise, as previous studies have shown that this discrepancy between observational and genetic analyses for elevated plasma levels was also present for the inflammatory biomarker CRP.11 Nevertheless, since plasma fibrinogen is widely associated with COPD-related outcomes,7 it has been speculated that it could be more usable than CRP as a biomarker in COPD due to a lower variability.38 Plasma fibrinogen has been suggested as a target for intervention and is currently considered for stratification in randomised controlled trials by the US FDA.8 ,9 ,39 The most reliable interpretations of genotype–disease associations are usually found when the polymorphism appears to directly influence the plasma levels such as for the β-fibrinogen genotype used in this study.22 As the fibrinogen polymorphism FGB -455 is located in the promoter region it affects plasma fibrinogen levels only without affecting protein structure.22
Plasma α1-antitrypsin is a positive acute phase protein,12 and several previous studies have corroborated that elevation of positive acute phase proteins is associated with increased risk of exacerbations in COPD. However, previous studies have also shown that plasma α1-antitrypsin lowered by the Z-allele is associated with increased risk of emphysema and thus COPD.14 ,40 ,41 Interestingly, we observed that elevation of the positive acute phase protein α1-antitrypsin is strongly associated with an increased risk of exacerbations in COPD. In contrast, we provide suggestive evidence that plasma α1-antitrypsin genetically lowered by the Z-allele is associated with an increased risk of exacerbations in individuals with COPD from the general population. It is important to note that Mendelian randomisation analyses are, indeed, based on strong assumptions, for example, that every pathway from a genotype to exacerbations pass through only one plasma marker.42 Further basic assumptions of Mendelian randomisation are that the genotype used as instrument is robustly associated with plasma levels and that the genotype is independent of possible confounding variables.37 As mentioned, we observed an association between α1-antitrypsin genotype and FEV1; however, adjusting our instrumental variable analyses for FEV1 only altered our results slightly. In logistic regression analyses the significant association between the Z-allele and increased odds of exacerbations was corroborated, and adjusting the logistic regression analysis for FEV1 did not affect our estimates. Interestingly, in our study we also observed that the plasma α1-antitrypsin-lowering S-allele was not associated with exacerbations. A possible mechanistic explanation could be that a Z-allele point mutation causes protein product polymers that increase the influx of neutrophils into the lungs, and this attribute of the Z-allele could increase the level of inflammation compared with the S-allele and, thereby, the risk of exacerbations.12 In support of this hypothesis, studies have shown that aggregation of intracellular Z α1-antitrypsin can cause a particularly severe cellular inflammatory phenotype in COPD.43
Limitations of our study include possible pleiotropic effects of the genotypes studied, implying that the genotypes have different effects on multiple organ systems such as the Z-allele associating with both airflow limitation and liver disease.44 Studies have shown that about one-third of individuals with the ZZ genotype develop clinical liver injury, and although the clinical presentation is variable,44 we cannot rule out that pleiotropic effects of the Z-allele could affect our findings; however, this is unlikely.
All individuals with the ZZ genotype were verified by control genotyping, and the method was validated by control sequencing. However, another possible limitation was that the α1-antitrypsin Z genotype deviated from the Hardy-Weinberg equilibrium. This reflects a lack of seven ZZ homozygotes in the population of more than 95 000 individuals with data on α1-antitrypsin genotype, possibly because they were too sick to participate in our study. This lack of a few ZZ individuals, though, would bias our estimates for the Z-carriers towards a null association, and is therefore unlikely to affect our conclusions. In addition, although we included extensive data on possible confounders, unmeasured confounding with common causes on genotypes and exacerbations could still be important in our study.42
Strengths of our study include the robust associations between genotypes and levels of all included plasma markers. We also believe that a strength of our study was the ability to include a CRP genotype located in the CRP promoter region combined with plasma CRP as a control in both observational and genetic analyses. Furthermore, the large number of individuals with COPD examined with pulmonary function tests in the Copenhagen General Population Study, with genotyping and measurements of plasma marker levels at the examination, in combination with assessment of exacerbations in all-inclusive nationwide registers without losses to follow-up, significantly increases the reliability of our results.
In conclusion, in this study we found that elevated plasma fibrinogen and elevated plasma α1-antitrypsin were associated with an increased risk of exacerbations in COPD. In contrast, it was genetically lowered plasma α1-antitrypsin that was associated with increased odds of exacerbations in COPD, while genetically elevated plasma fibrinogen was not, indicating that fibrinogen per se is not causally associated with exacerbations while α1-antitrypsin is.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors Study concept and design: TSI, LR, JV, BGN; acquisition of data: PL, BGN; analysis and interpretation of data: TSI, LR, JLM, JV, BGN; first drafting of the manuscript: TSI; critical revision of the manuscript for important intellectual content: all authors; statistical analysis: TSI, LR, JLM; obtained funding: PL, JV, BGN; administrative, technical and material support: BGN, PL; study supervision: LR, JV, BGN; data access and responsibility: TSI had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis and for the submission.
Funding The Copenhagen General Population Study is supported by the Capital Region of Copenhagen, the Danish Heart Foundation, the Danish Lung Foundation, the Velux Foundation and Herlev Hospital.
Competing interests JV has received honoraria from GlaxoSmithKline, Almirall, AstraZeneca, Boehringer-Ingelheim, Novartis and Takeda for consulting and for presenting at meetings and symposia. PL has received honoraria from GlaxoSmithKline and other pharmaceutical companies for consulting, teaching and presenting at meetings and symposia.
Ethics approval The study was approved by Herlev Hospital and a Danish regional ethics committee (H-KF01-144/01) and was conducted according to the Declaration of Helsinki. Written informed consent was obtained from all individuals.
Provenance and peer review Not commissioned; internally peer reviewed.
Linked Articles
- Airwaves