Article Text

Abstract
Background Chronic obstructive pulmonary disease (COPD) is a progressive, incurable lung disease characterised by abnormal tissue repair causing emphysema and small airways fibrosis. Since current therapy cannot modify this abnormal repair, it is crucial to unravel its underlying molecular mechanisms. Unbiased analysis of genome-wide gene expression profiles in lung tissue provides a powerful tool to investigate this.
Methods We performed genome-wide gene expression profiling in 581 lung tissue samples from current and ex-smokers with (n=311) and without COPD (n=270). Subsequently, quantitative PCR, western blot and immunohistochemical analyses were performed to validate our main findings.
Results 112 genes were found to be upregulated in patients with COPD compared with controls, whereas 61 genes were downregulated. Among the most upregulated genes were fibulin-5 (FBLN5), elastin (ELN), latent transforming growth factor β binding protein 2 (LTBP2) and microfibrillar associated protein 4 (MFAP4), all implicated in elastogenesis. Our gene expression findings were validated at mRNA and protein level. We demonstrated higher ELN gene expression in COPD lung tissue and similar trends for FBLN5 and MFAP4, and negative correlations with lung function. FBLN5 protein levels were increased in COPD lung tissue and cleaved, possibly non-functional FBLN5 protein was present. Strong coexpression of FBLN5, ELN, LTBP2 and MFAP4 in lung tissue and in silico analysis indicated cofunctionality of these genes. Finally, colocalisation of FBLN5, MFAP4 and LTBP2 with elastic fibres was demonstrated in lung tissue.
Conclusions We identified a clear gene signature for elastogenesis in COPD and propose FBLN5 as a novel player in tissue repair in COPD.
- COPD ÀÜ Mechanisms
- Emphysema
Statistics from Altmetric.com
Key messages
What is the key question?
-
Which relevant lung tissue gene expression changes can explain the mechanisms behind abnormal tissue repair in chronic obstructive pulmonary disease (COPD)?
What is the bottom line?
-
Using genome-wide gene expression profiling in a large number of lung tissue samples from patients with COPD and non-COPD controls, we identified a clear COPD gene signature with increased expression of genes related to elastogenesis, with fibulin-5 as a main player.
Why read on?
-
Our findings provide much needed insights in lung tissue gene expression changes in COPD and point out fibulin-5 as a potential novel player in tissue repair in COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic and progressive lung disease with a large impact on patients and society and is mainly caused by cigarette smoking. There is no cure available for COPD and current drugs are mainly effective in improving symptoms and exacerbations but generally do not slow down the progression of the disease. Quitting smoking can reduce the accelerated lung function decline in some but not all patients with COPD and it does not restore lost lung function.
Pathologically, COPD is characterised by distinct features, including emphysema with the destruction of alveolar lung tissue and fibrosis of the (small) airways with airway wall thickening.1 Although emphysema and airway wall thickening occur in close proximity to each other, they seem to be driven by different processes since emphysematous destruction is characterised by insufficient extracellular matrix (ECM) production, indicating a defective repair process, whereas airway wall thickening is the result of an exaggerated repair process accompanied by a marked increase in ECM. Both processes are important contributors to loss of lung function in patients with COPD. Hogg and colleagues recently demonstrated that small airway wall thickening and loss of small airways precedes emphysematous lung tissue destruction in patients with COPD, suggesting a relation between these processes.2
The exact nature of the abnormal repair processes in COPD lungs is unknown, hence there is no effective treatment to prevent further lung tissue damage or induce repair. A better understanding of regulation of the repair processes and the underlying molecular mechanisms is thus crucial.
Unbiased analysis of genome-wide gene expression in lung tissue provides a powerful tool to investigate these molecular mechanisms. However, studies using this approach have been hindered thus far by small sample sizes due to the limited availability of lung tissue samples.3–7 As part of a Lung eQTL Consortium,8–10 we have performed an unbiased analysis of genome-wide gene-expression profiles in lung tissue specimens derived from a large number (N=581) of well characterised current and ex-smokers with (N=311) and without (N=270) COPD.
Methods
Detailed methods can be found in the online supplement.
Patients
Gene expression data were used from 311 patients with COPD and 270 non-COPD controls who were part of the Lung eQTL dataset from three academic sites.8 ,9 Lung tissue samples were collected from patients undergoing lung surgery for various reasons, mostly therapeutic resection of lung tumours, but also lung transplantation. In case of tumour resections, macroscopically normal lung tissue was taken far distant from the tumour and histology of all samples was checked for abnormalities using standard haematoxylin and eosin staining. Lung samples were obtained in accordance with local ethical guidelines (for a detailed description, see Hao et al8). In the current analyses, we included current and ex-smokers >40 years with ≥5 pack-years. COPD was defined as an FEV1/FVC ratio <70%. Non-COPD control was defined as an FEV1/FVC ≥ 70% predicted. In case lung tissue samples were derived from healthy donors, no data on FEV1 or FEV1/FVC ratio were available. For FEV1 and FEV1/FVC, pre-bronchodilator values were used when post-bronchodilator values were not available. Subjects with other lung diseases such as asthma, cystic fibrosis or interstitial lung diseases were excluded.
Gene expression assays
mRNA profiling was performed by Rosetta Inpharmatics Gene Expression Laboratory (Seattle, Washington, USA) using custom Affymetrix HU133 arrays (GEO platform GPL10379), consisting of 751 control and 51 627 non-control probe sets as described previously.8 Gene expression normalisation was performed using Robust Multi-array Average and every probe set was normalised; that is, the mean expression was set to zero and the SD to one. We corrected for strong expression differences between the lung tissue samples due to unknown factors (eg, batch or technical effects) by correcting for the first 25 principal components, as described previously.11
We analysed the three cohorts separately, and corrected for potential confounders: age, gender, pack-years and smoking status (current vs former). We performed t tests to find differentially expressed genes between patients with COPD and controls. We then used an inverse variance meta-analysis to identify genes that were differentially expressed and behaved in the same direction across all three cohorts. We used the false discovery rate (FDR) to control for multiple testing. We report on a set of differentially expressed genes of which we accepted that 20% might be false positives. We observed that a nominal p<0.001 resulted in 252 differentially expressed genes in the real analysis, whereas we would expect 51.6 by chance, corresponding to an FDR of 0.20.
GeneNetwork analysis
A new method was used to gain insight into the potential gene function of differentially expressed genes, that is, GeneNetwork. This method uses an independent gene expression dataset of 77 840 samples to predict the function of genes in an unbiased way, as recently employed.12 We used this method to predict (currently unknown) gene functions based on known biological pathways available in the molecular signatures database MSigDB (http://www.broadinstitute.org) and additionally used this information for pathway enrichment analyses and cofunctionality networks. Knockout information from the Mouse Genome Informatics database was collected for genes for which human orthologues exist to predict phenotypes in mice for which currently no knockout has been described.
Quantitative PCR validation
Quantitative PCR (qPCR) was used to confirm the expression levels of ELN (elastin), FBLN5 (fibulin-5), MFAP4 (microfibrillar associated protein) and LTBP2 (latent transforming growth factor (TGF)-β binding protein 2) in lung tissue samples from the Groningen cohort.
Additionally, FBLN5 expression was analysed in primary pulmonary fibroblasts treated with or without TGFβ and cigarette smoke extract (CSE) from patients with stage IV COPD and non-COPD controls.
Histology
Immunohistochemical staining for FBLN5, MFAP4 and LTBP2 was performed on paraffin embedded lung tissue samples from the Groningen cohort to localise protein expression in lung tissue and assess the presence of these proteins in elastic fibres.
Western blot analysis
Western blot analyses was performed to measure total protein levels of FBLN5, ELN and MFAP4 in lung tissue and to identify the presence of the cleaved form of FBLN5.
Statistics
Gene expression analyses were performed using the eQTLMappingPipeline as previously described.13 Additional statistical analyses were performed using IBM SPSS Statistics 22 and R statistical software V.2.12.0. Differences in clinical characteristics were analysed using Mann–Whitney U (MWU) tests. For qPCR validation, MWU tests were used and increased gene expression and a one-sided p value <0.05 was considered a positive validation. Differences in FBLN5 expression levels in COPD and control fibroblasts and differences in FBLN5 protein levels in lung tissue were assessed using MWU tests and a two-sided p value <0.05 was considered significant. The relation between FEV1 and FBLN5, ELN, LTBP2 and MFAP4 expression and the relation between mRNA expression of these four genes in lung tissue was assessed by Spearman's correlation. A p<0.05 was considered significant.
Results
Subject characteristics
Table 1 presents the clinical characteristics of the subjects. We included 146 subjects from the Groningen cohort, 282 from the Laval cohort and 153 from the UBC cohort. The subjects from the Groningen cohort were somewhat younger than those from Laval and UBC, had less pack-years smoked and had more severe COPD. Overall, patients with COPD and non-COPD controls did not differ in age, gender and smoking status, but patients with COPD had more pack-years smoked than the non-COPD controls. Since these phenotypic differences exist between the cohorts we performed subsequent analyses per cohort separately, followed by a meta-analysis.
Subject characteristics
Differentially expressed genes in COPD
The expression of 171 probe sets corresponding to 112 known genes was upregulated in patients with COPD versus non-COPD controls, and the expression of 81 probe sets corresponding to 61 known genes was downregulated at a p value <0.001, corresponding to an FDR of 20%. A heatmap of these findings is shown in figure 1. The complete list of all probe sets and statistics is shown in online supplementary table S1. Among the most upregulated genes were FBLN5, microtubule-actin cross linking factor 1 (MACF1) and ELN. Among the most downregulated genes were immunoglobulin heavy chain constant region μ (IGHM), vascular endothelial growth factor α (VEGFA) and periostin (POSTN).
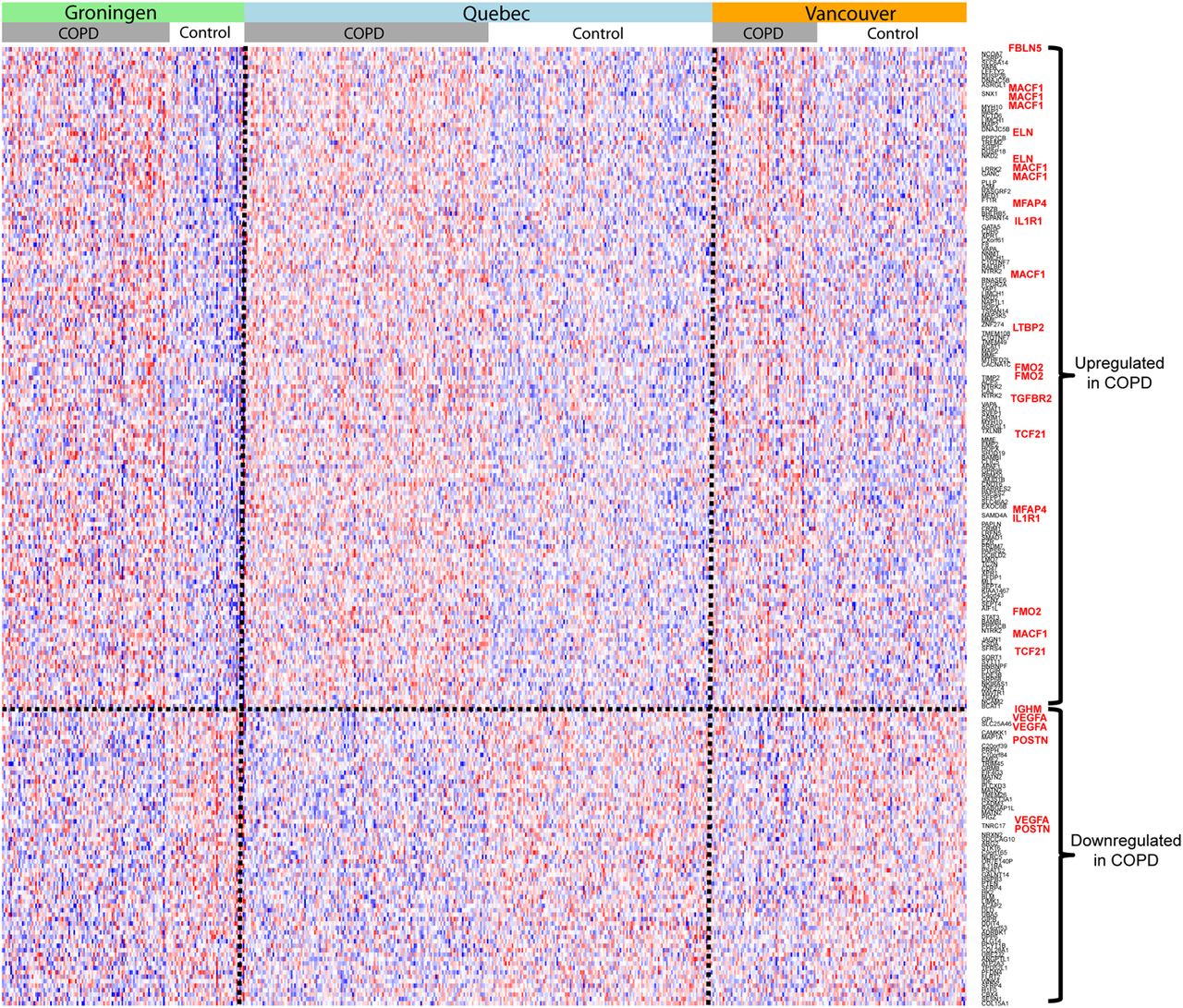
Heatmap of differentially expressed genes in chronic obstructive pulmonary disease (COPD). The heatmap shows differential gene expression (p<0.001) between COPD and non-COPD control lung tissue for the three patients cohorts from Groningen, Quebec and Vancouver. Each probe set is shown separately and for several genes we found more than one significant probe set. The coloured, grey and white bars above the heatmap indicate the different groups of lung tissue samples. Genes with a higher expression in COPD lung tissue are shown in the upper part of the heatmap, genes with a lower expression in the lower part. Blue represents lower and red higher relative gene expression. The genes most relevant for this manuscript are highlighted in red on the left side of the heatmap.
From the upregulated genes, FBLN5 is most interesting in the context of COPD pathogenesis. This gene has been shown to be essential for elastic fibre formation, it is abundantly expressed during lung development,14 and FBLN5 knockout mice were shown to develop severe emphysema.15 ,16 Moreover, three additional genes implicated in elastogenesis were found to be among the most upregulated genes in COPD, that is, ELN, LTBP2 and MFAP4.17 ,18
Other upregulated genes considered to be of potential interest for COPD pathogenesis are TGFβ receptor-2 (TGFBR2), α-2-macroglobulin (A2M), interleukin 1 receptor type 1 (IL1R1), transcription factor 21 (TCF21) and metallopeptidase inhibitor 2 (TIMP2).
Given the potential novel roles for FBLN5, LTBP2 and MFAP4 in elastogenesis in COPD, we decided to further focus on these genes in this article.
Pathways and phenotypes related to (abnormal) lung development and emphysema are enriched among genes upregulated in COPD
We subsequently assessed the enrichment of biological processes and predicted phenotypes among the 112 genes upregulated in patients with COPD at p<0.001. While such an analysis is typically conducted using gene set enrichment analysis (GSEA), we performed a similar analysis but instead of using only established gene functions in GSEA, we used the predicted gene functions that were based on the independent gene expression dataset of 77 840 samples.
Enrichment was found for biological processes involved in lung development, cell adhesion and fibroblast migration, and several phenotypes related to abnormal lung development and lung morphology and emphysema (table 2).
Pathway enrichment based on gene function prediction of genes upregulated in COPD
Overlapping gene function and cofunctionality for elastogenesis genes
To gain more insight in the function of the four genes related to elastogenesis, we used our GeneNetwork method to predict potential functions of genes (see online supplementary methods). We found a striking overlap in predicted gene function for the four elastogenesis genes, most being predicted to be involved in ECM organisation, collagen fibril organisation, cell-matrix adhesion, and the TGFβ receptor signalling pathway (table 3A). Interestingly, we also found that all four genes related to elastogenesis were predicted to give an emphysematous phenotype in knockout mice (table 3B). Other predicted phenotypes in knockout mice were overexpanded pulmonary alveoli, impaired lung alveolar development and dilated airways.
Gene function prediction in biological pathways
Gene function prediction in mouse knockout phenotypes
When assessing which genes share many predicted functions, we found a very consistent gene network for the genes upregulated in COPD at p<0.001. The four genes related to elastogenesis (FBLN5, ELN, MFAP4 and LTBP2) clearly cluster together and are surrounded by other genes relevant for COPD pathogenesis (figure 2). The gene network for the downregulated genes was less consistent (see online supplemental figure S1).

Cofunctionality network of genes upregulated in patients with chronic obstructive pulmonary disease (COPD). The cofunctionality network shows the clustering of all genes that are upregulated in COPD lung tissue at p<0.001. The clustering is based on the overlap in gene function as predicted by our GeneNetwork; that is, close clustering means high overlap in predicted gene function. The four elastogenesis genes are highlighted in dark pink and cluster together in the centre of the network. Several other genes relevant for COPD pathogenesis are highlighted in light pink.
Validation of gene expression findings using qPCR
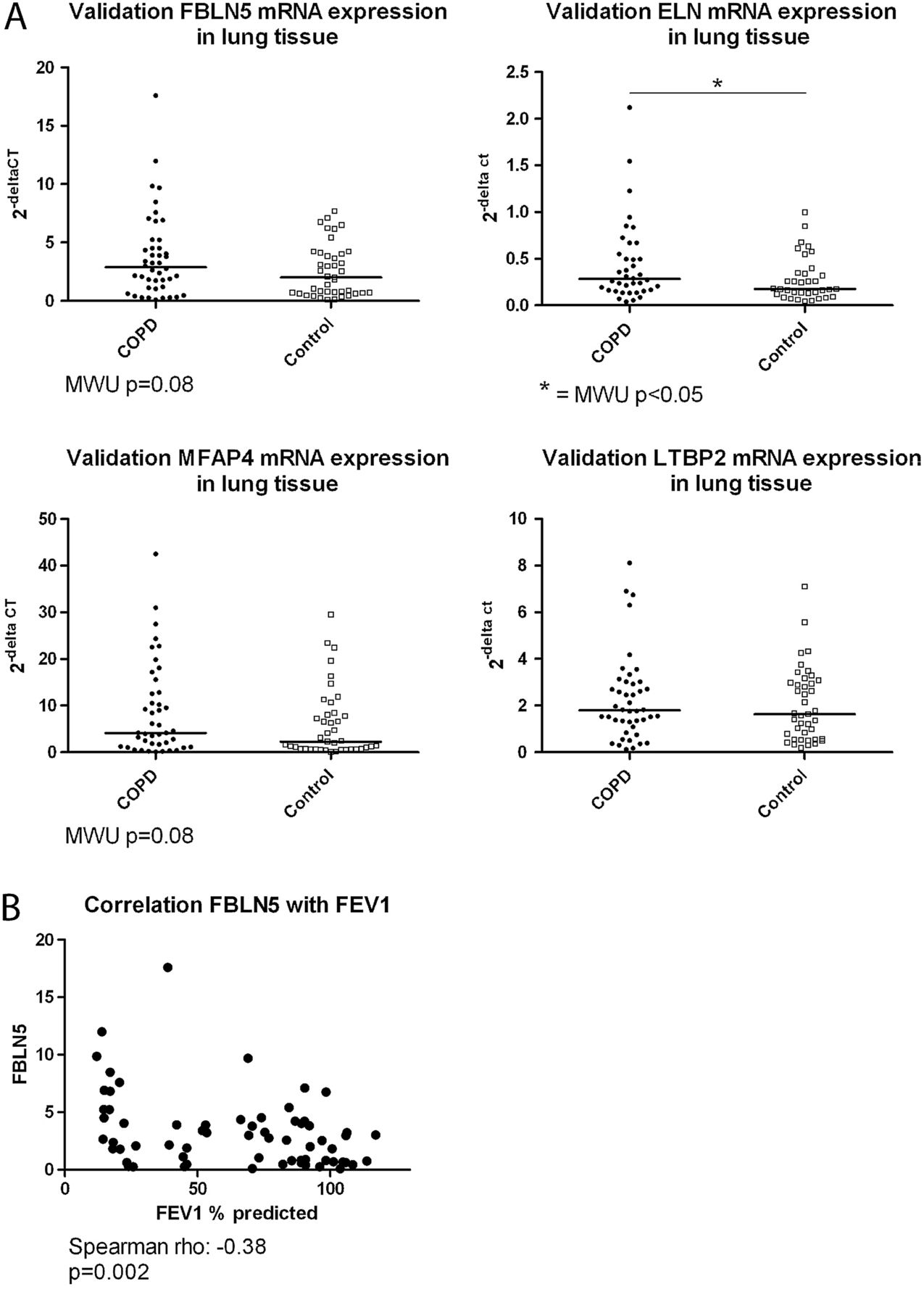
qPCR for FBLN5, ELN, MFAP4 and LTBP2 was performed on lung tissue samples from 45 patients with COPD and 42 non-COPD controls from the Groningen cohort. Subject characteristics are given in online supplementary table S2.
We found significantly higher ELN expression in COPD compared with control lung tissue and similar trends for FBLN5 (p=0.08) and MFAP4 (p=0.08) (figure 3A). There was no difference in LTBP2 expression. In addition, significant negative correlations were found between FEV1 and FBLN5 (figure 3B), and FEV1 and ELN, and FEV1 and MFAP4 (see online supplemental figure S2A). Moreover, strong coexpression was observed between these four elastogenesis genes (see online supplemental figure S2B), which shows the robustness of our findings and validates the similarity in the predicted function for these four genes.
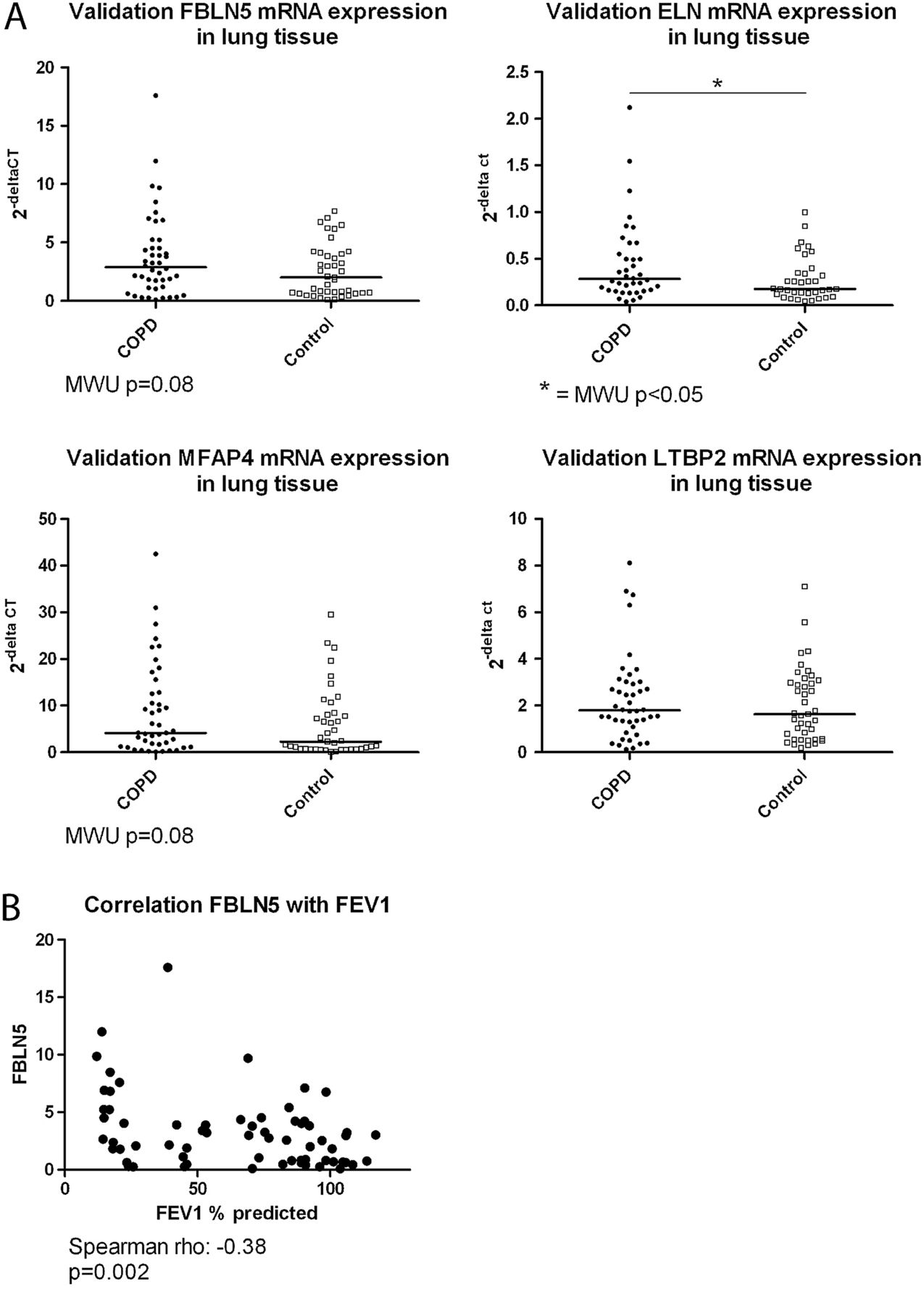
Quantitative PCR validation. (A) Fibulin-5 (FBLN5), elastin (ELN), microfibrillar associated protein 4 (MFAP4) and latent transforming growth factor β binding protein 2 (LTBP2) mRNA expression relative to housekeeping gene expression in lung tissue comparing patients with chronic obstructive pulmonary disease (COPD) (closed symbols) and non-COPD controls (open symbols). The results of the Mann–Whitney U (MWU) analyses are depicted in the figure. (B) The negative correlation between the expression of FBLN5 in lung tissue and FEV1 % predicted is shown. The result of the Spearman correlation is depicted below the figure.
FBLN5 is expressed in primary pulmonary fibroblasts and strongly induced by TGFβ
As pulmonary fibroblasts are the main source of ECM proteins in the lung, and thus important for elastic fibre formation, we investigated FBLN5 gene expression levels in primary pulmonary fibroblasts derived from patients with COPD and non-COPD controls from the Groningen cohort. To this end, fibroblasts were treated with or without TGFβ and CSE (figure 4). Under basal conditions there was no significant difference in FBLN5 expression between COPD and non-COPD control fibroblasts, although COPD fibroblasts tended to have higher expression. However, there was a clear increase in FBLN5 expression after TGFβ treatment in pulmonary fibroblasts, indicating a repair response. CSE exposure did not affect FBLN5 expression in pulmonary fibroblasts.
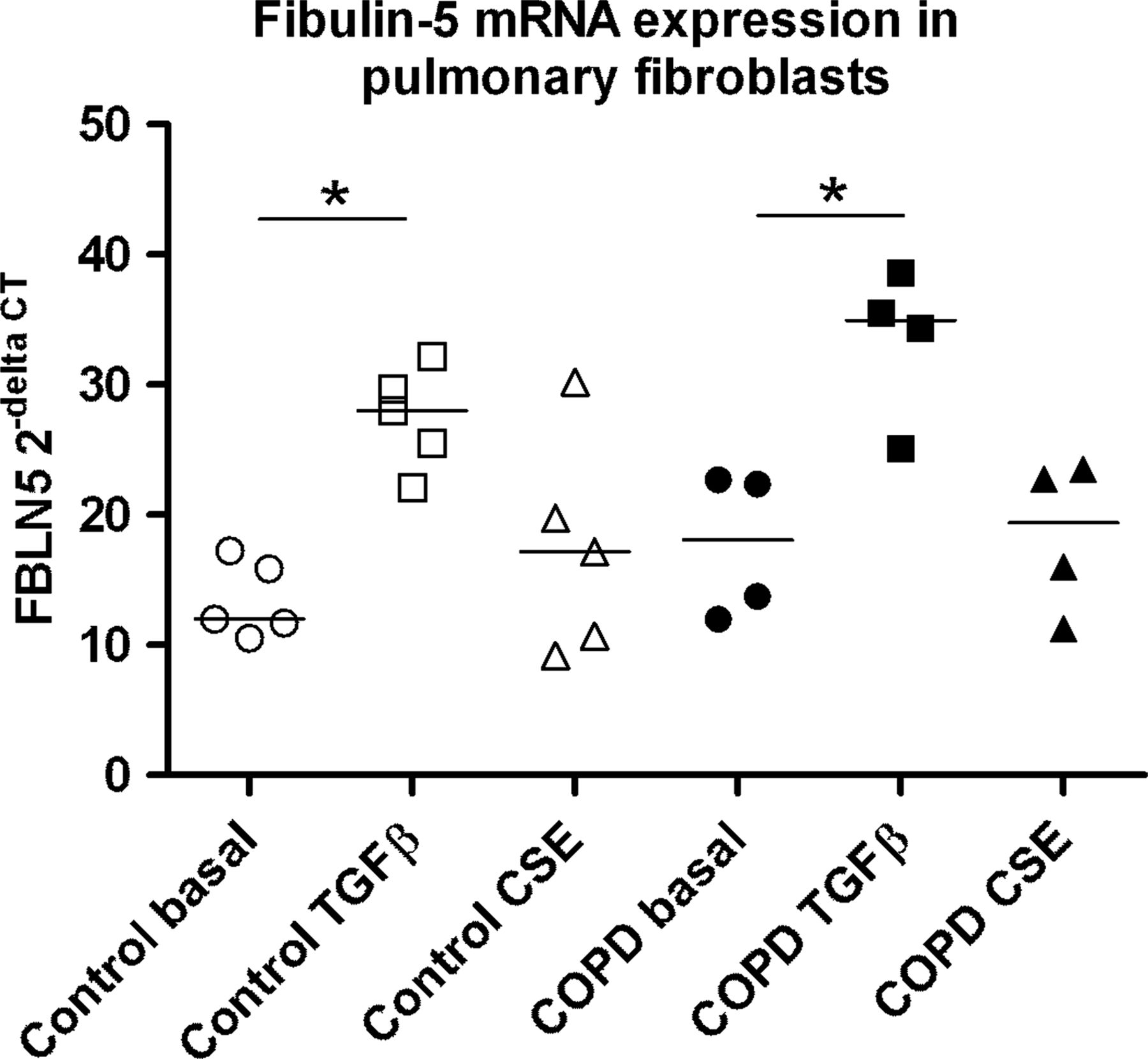
Fibulin-5 (FBLN5) expression in pulmonary fibroblasts. FBLN5 gene expression in pulmonary fibroblasts from control subjects (open symbols. n=5) and patients with chronic obstructive pulmonary disease (COPD) (closed symbols, n=4) is shown during basal conditions (circles) and after transforming growth factor β (TGFβ) (squares) and cigarette smoke extract (CSE) (triangles) treatment. Differences between the groups were tested by Mann–Whitney U tests. *p<0.05.
FBLN5, MFAP4 and LTBP2 are localised in elastic fibres in the lung
Immunohistochemistry was performed on serial cut lung tissue sections to assess colocalisation of FBLN5, MFAP4 and LTBP2 with elastic fibres in the lung. Figure 5 shows clear staining of FBLN5 in alveolar and vessel walls, and surrounding the airways, which was similar in localisation to elastin. LTBP2 and MFAP4 staining was present in vessel walls and the matrix surrounding airways and vessels. LTBP2 and MFAP4 were also present in areas containing elastic fibres.
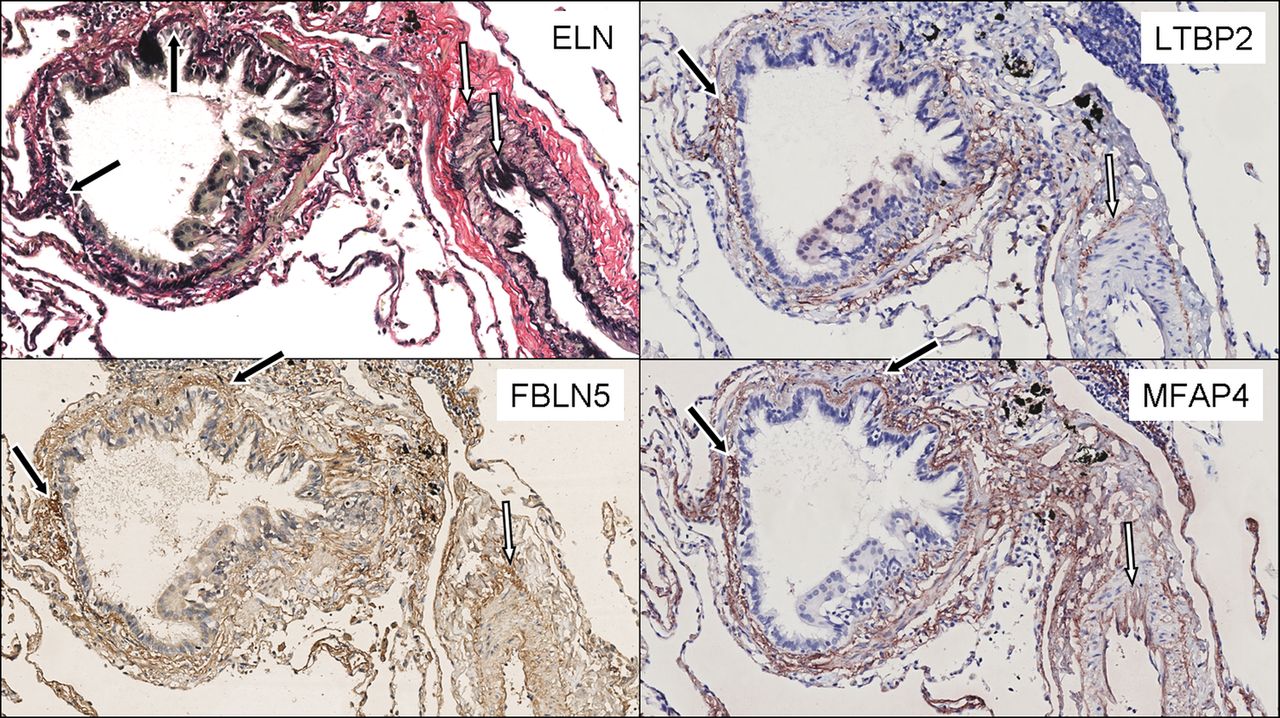
Elastin (ELN), fibulin-5 (FBLN5), latent transforming growth factor β binding protein 2 (LTBP2) and microfibrillar associated protein 4 (MFAP4) staining in lung tissue. Presence of elastic fibres (black) is demonstrated with a Verhoeff van Gieson stain in the upper left panel. LTBP2 and MFAP4 staining (red) is shown in the right upper and lower panel, respectively. FBLN5 staining (brown) is shown in the lower left panel. Examples of colocalisation with elastic fibres are indicated with black arrows in the matrix around the airways and white arrows in the vessel walls.
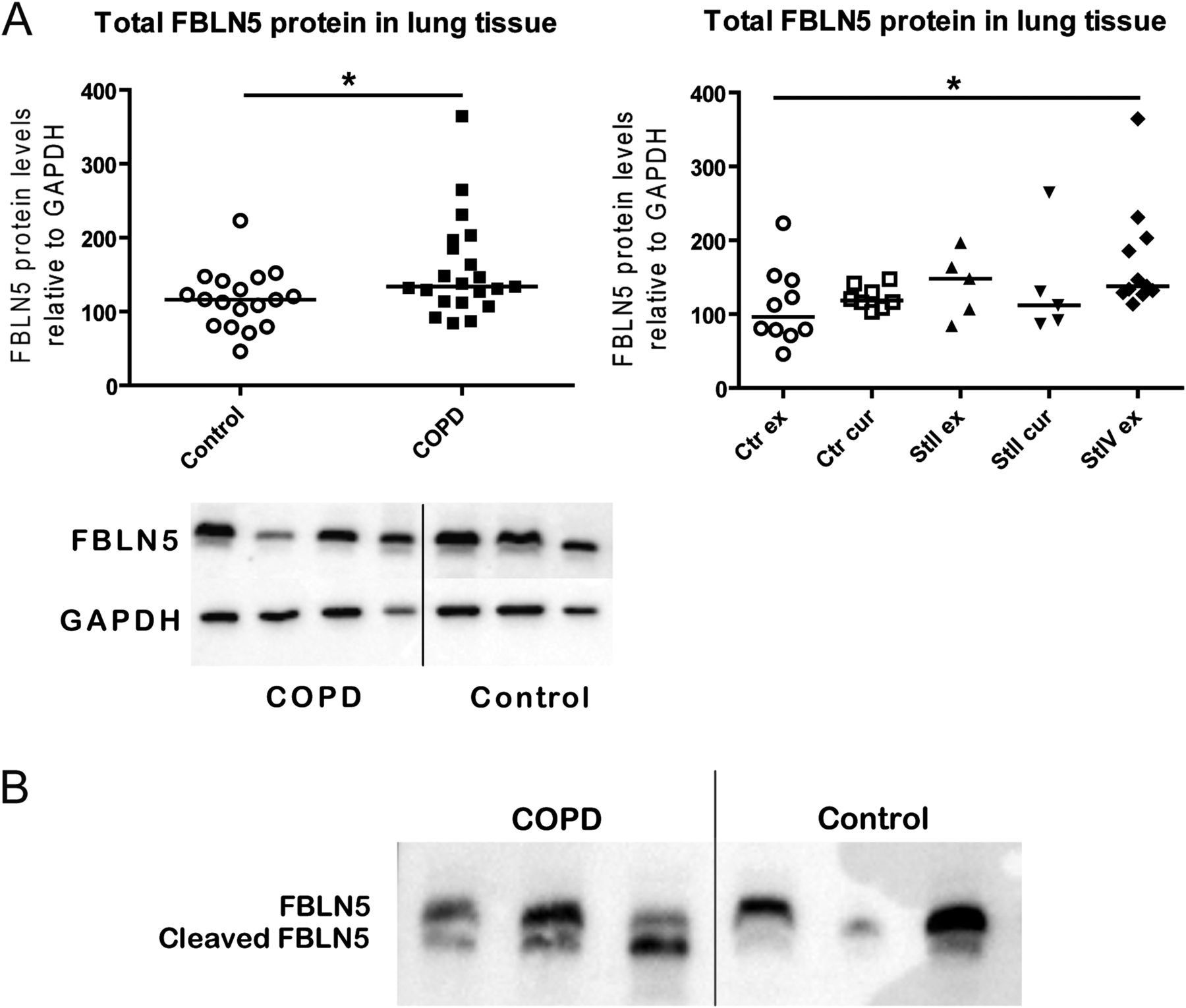
Increased FBLN5 protein levels in COPD lung tissue
FBLN5, ELN and MFAP4 protein levels were analysed in lung tissue samples from 24 patients with COPD and 19 non-COPD controls from the Groningen cohort (see online supplementary table S3) by western blot.
Total levels of FBLN5 protein were significantly higher in COPD than control lung tissue (figure 6A), an effect most clearly seen when comparing control ex-smokers and COPD stage IV ex-smokers. We also demonstrated the presence of cleaved FBLN5 protein in lung tissue and found higher levels in COPD compared with control lung tissue (figure 6B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Total and cleaved fibulin-5 (FBLN5) protein levels in lung tissue. (A) FBLN5 protein levels relative to GAPDH expression in lung tissue comparing patients with chronic obstructive pulmonary disease (COPD) and non-COPD controls. The left graph shows the comparison between all patients with COPD (closed symbols) and controls (open symbols). The right graph shows the subgroups based on smoking status and COPD stages. A representative western blot of FBLN5 and GAPDH is shown below the graphs. Differences between the groups were tested by Mann–Whitney U tests. *p<0.05. (B) Western blot staining showing total FBLN5 and cleaved FBLN5 protein levels in lung tissue of three patients with COPD and three non-COPD controls. GADPH, glyceraldehyde 3-phosphate dehydrogenase.
There were no significant differences in total protein levels of ELN and MFAP4 between COPD and control lung tissue (see online supplemental figure S3), yet ELN protein levels were lower in current than ex-smoking controls.
Discussion
Using genome-wide gene expression analyses on a large dataset of lung tissue specimens from patients with COPD and non-COPD controls, we identified a clear gene signature for elastogenesis in COPD, with increased expression of ELN, FBLN5, MFAP4 and LTBP2. Among the upregulated genes in COPD, we demonstrated a clear enrichment of genes that are known to induce an emphysematous phenotype if knocked out in mice, including FBLN5. Additionally, we validated part of our gene expression findings using qPCR and showed a negative correlation between FBLN5, ELN and MFAP4 expression and lung function. We showed strong correlations between the gene expression of ELN, FBLN5, MFAP4 and LTBP2 and we observed colocalisation of FBLN5, MFAP4 and LTBP2 protein with elastic fibres in lung tissue. Thus the presence of these elastogenesis genes and proteins is strongly related in lung tissue, in line with the overlap in gene function that we predicted using our independent gene expression dataset. Finally, we demonstrated higher protein levels of total FBLN5 and cleaved FBLN5 protein in lung tissue from patients with COPD than non-COPD controls.
The gene that was most significantly upregulated in COPD, and most interesting with respect to COPD pathogenesis, was FBLN5. FBLN5 is essential for elastic fibre assembly and abundantly expressed during lung and vascular development.14 FBLN5 knockout mice survive to adulthood but develop pronounced elastinopathy with human aging phenotypes like loose skin, vascular abnormalities, severe emphysema and genital prolapse.15 ,16 This phenotype in mice is very similar to the human connective tissue disorder cutis laxa, which can be caused by mutations in FBLN5 and is often associated with severe emphysema.19 Based on these findings we consider FBLN5 as a potential novel player in tissue repair in COPD and decided to further focus on this gene. Next to FBLN5 and ELN itself, two other genes related to elastogenesis, that is, MFAP4 and LTBP2, were also upregulated in COPD. MFAP4 colocalises with elastin in the lung18 and LTBP2 has been shown to interact with FBLN5 and promote elastic fibre assembly.17
Interestingly, FBLN5, LTBP2 and MFAP4 were present in elastic fibres in all lung compartments, hence they could potentially be involved in vascular and matrix related changes associated with emphysema and COPD, which may be relevant for further studies.
Our findings are intriguing yet paradoxical as elastic fibres are destroyed with emphysema, resulting in severely impaired lung elasticity in COPD. We propose that upregulation of these elastogenesis genes in COPD is the result of an attempt to repair the damaged lung, an attempt that is not effective. This hypothesis is supported by the observation that FBLN5 gene expression increases after elastase-induced emphysema in mice,20 and is also in line with previous studies demonstrating increased elastin gene expression in severe COPD.21 Furthermore, our results and those of Kuang et al,22 showing increased FBLN5 gene expression after TGFβ treatment in lung fibroblasts, support a role for FBLN5 in repair responses.
A possible explanation for the fact that this repair response in COPD is not effective might be found in defects in protein translation. Two studies have now demonstrated that the FBLN5 protein can be cleaved by serine proteases23 ,24 and increased levels of this cleaved form of FBLN5 protein were demonstrated in the skin of aged mice.23 This is intriguing as on the one hand COPD has been regarded as an aging lung disease25 ,26 and on the other hand increased serine protease activity is present in COPD lungs, which is thought to play an important role in emphysematous lung tissue destruction.27 ,28 Moreover, the cleaved form of FBLN5 does not promote elastic fibre assembly in vitro, and thus seems to be non-functional.23 To investigate the role of cleaved FBLN5, we measured its protein expression in lung tissue and found higher levels in patients with COPD than in non-COPD controls.
In addition to the four genes related to elastogenesis, we found several other genes to be upregulated in COPD lung tissue that might be relevant for COPD pathogenesis. MACF1 is involved in the actin cytoskeleton organisation and was demonstrated to be a positive regulator of WNT signalling,29 an important lung developmental pathway.30 TGFBR2 is one of the receptors for TGFβ, which is involved in many cellular processes including lung development and wound repair and often implicated in COPD.31 TCF21 is involved in epithelial–mesenchymal interactions in kidney and lung morphogenesis and is important in epithelial differentiation and branching morphogenesis.32 α2-Macroglobulin (A2M) is a protease inhibitor and a genetic defect in this gene has been associated with COPD.33
Among the downregulated genes in COPD lung tissue, VEGFA and POSTN were most relevant for COPD pathogenesis. VEGF is an important stimulator of angiogenesis and decreased VEGF levels have been associated with emphysema in humans and in mice.34 POSTN is a matricellular protein that has been associated with T helper 2 related subepithelial fibrosis in asthma35 and progression of fibrosis in patients with idiopathic pulmonary fibrosis.36
A strength of our study was the very stringent way of analysing gene expression data. To identify the differentially expressed genes we corrected for age, gender, smoking status and pack-years and studied each of the three cohorts separately and subsequently conducted a meta-analysis. However, we also corrected the expression data for the 25 most dominant expression effects (identified by principal component analysis). One reason to do this is that some of these components might capture unknown, confounding factors that otherwise could result in false positive associations. While this approach invariably will result in false negative findings, the small set of genes that we report on here showed a highly consistent expression difference between cases and controls that was present in each of the three cohorts, showing the robustness of our findings. Opposed to our stringent approach we decided to use a lenient FDR cut-off of 20% to assess the differentially expressed genes. This resulted in 252 differentially expressed genes as opposed to 51.6 that are expected by chance. The FDR was estimated very conservatively, assuming that the 52 738 tests (ie, each probe present on the expression platform) were entirely uncorrelated. In reality, this is not the case, since many probes (especially those mapping within the same gene) typically show strong coexpression. As such we feel confident that the actual FDR of these 252 differentially expressed probes is lower than 20%. We choose this FDR as a hypothesis generating cut-off, and this might give some false positive results. Therefore we performed several follow-up experiments and validated our main findings on mRNA and protein level, demonstrated colocalisation with elastic fibres in lung tissue and showed cofunctionality between the upregulated genes.
We used a novel method that can accurately predict gene function based on an independent gene expression dataset of 77 840 samples. A clear overlap in predicted gene function was found for the genes related to elastogenesis and for the other upregulated genes which are relevant for COPD pathogenesis. Visualisation in a cofunctionality network showed that these genes share many functions. Moreover, among the genes upregulated in COPD, enrichment was found for genes that are predicted to give an emphysematous phenotype in knockout mice, confirming that our results are indeed specific for COPD.
In conclusion, we identified a clear elastogenesis gene signature in COPD, with increased gene expression of ELN, FBLN5, MFAP4 and LTBP2 in lung tissue. We provide evidence for FBLN5 as a novel player in abnormal tissue repair in COPD. We suggest that increased FBLN5 mRNA expression in COPD lungs is due to a response to tissue damage and repair, and that cleavage of FBLN5 protein is increased in COPD lungs due to high serine protease activity. Cleaved FBLN5 is non-functional and does not contribute to elastic fibre assembly and tissue repair in COPD. Functional studies are currently ongoing to unravel the exact role of FBLN5 in tissue repair in COPD.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tableS1
Footnotes
-
Contributions C-AB was involved in lung tissue collection and compiling the lung tissue database, contributed to the study concept and design, interpreted the data, supervised the basic laboratory research and drafted the manuscript. MvdB was involved in compiling the lung tissue database and gathering of clinical data, contributed to the study concept and design, was involved in data analyses and data interpretation, and writing of the manuscript. DSP, WT, PDP, DDS, YB, ML contributed to the study concept and design, were involved in lung tissue collection and compiling the lung tissue database, including clinical information, and the writing and final editing of the manuscript. In addition, DSP and WT contributed to the data interpretation. DCN and KH were involved in the study concept and design, edited the final manuscript and were responsible for the gene expression profiling. MRJ, AIRS and SB conducted the basic laboratory research and contributed to the writing and final editing of the manuscript. JK and RSNF developed the GeneNetwork under supervision of LF. LF had full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analyses. In addition, he contributed to the writing and final editing of the manuscript. C-AB and MvdB are co-first authors; WT and LF are co-last authors.
-
Funding This study was funded by the Dutch Lung Foundation (3.2.11.024) and Merck Research Laboratories.
-
Competing interests None.
-
Ethics approval Ethics committees of the Institut universitaire de cardiologie et de pneumologie de Québec (Quebec cohort) and the UBC-Providence Health Care Research Institute Ethics Board (Vancouver cohort).
-
Provenance and peer review Not commissioned; externally peer reviewed.